
Iron-catalyzed cyanoalkylation of difluoroenol silyl ethers with cyclobutanone oxime esters
2022-06-18 03:00XioleiZhuYngenHungXiuhuXuFenglingQing
Chinese Chemical Letters 2022年2期
Xiolei Zhu, Yngen Hung, Xiuhu Xu, Fengling Qing,
a College of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai 201620, China
b Key Laboratory of Organofluorine Chemistry, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Science, Chinese Academy of Science, Shanghai 200032, China
ABSTRACT An iron-catalyzed coupling reaction of difluoroenol silyl ethers and cyclobutanone oxime esters is described.This protocol provides a convenient access to various previously unknown and potentially useful gem-difluoromethylenated ketonitriles inmoderate to good yields.The transformations of resulting products to other fluorinecontaining products is also documented.
Keywords:Cyanoalkylation Difluoroenol silyl ether Cyclobutanone oxime ester Iron catalysis Radical reaction
The introduction of agem-difluoromethylene (CF2) unit into organic molecules often dramatically perturbs their physical, chemical and biological properties [1–3].For instance, when the CF2group was introduced adjacent to the carbonyl functionality, the electrophilicity of the carbonyl carbon atom is sharply increased.This causes not only changes in reactivity but in biological activity as well.It is well known thatα,α-difluoroketones tend to exist in hydrated form and mimic closely the tetrahedral transition state,which has been applied in the development of enzyme inhibitors(Fig.1.) [4,5].Furthermore,α,α-difluoroketones are valuable building blocks for the preparation of various useful fluorine-containing compounds [6–9].
Over the past decades, promising strategies have been reported for the synthesis ofα,α-difluoroketones, mainly including fluorination of ketones [10–14], oxidation ofα,α-difluoroalcohols [15–19], and transformation of difluorocarbonyl synthons [20–24].The fluorination methods have some drawbacks such as poor functional compatibility, whereas oxidation protocol always requires prefunctionalized substrates.In contrast, with the increasing accessibility of difluorocarbonyl synthons, considerable efforts have been devoted to their conversions toα,α-difluoroketones.Among the difluorocarbonyl synthons, difluoroenol silyl ethers have recently received much attentions due to their versatile reactivity[25,26].They are easily prepared from trifluoromethylketones by Mg-mediated cleavage of one of the C-F bonds [27,28].The standard reactions of difluoroenol silyl ethers involve nucleophilic addition to unsaturated substrates (Scheme 1a) [29–33] and substitution of electrophiles (Scheme 1b) [34–38].In 2009, our group reported an unique copper-catalyzed oxidative coupling of difluoroenol silyl ethers with tertiary amines to accessβ-amino-α,αdifluoro ketones (Scheme 1c) [39].Recently, the reactions of difluoroenol silyl ethers and different radical precursors, such as aryl diazonium salts, Katritzky salts, and alkyl carboxylic redox esters have been achieved under photoredox catalysis (Scheme 1d) [40–43].Despite these elegant advances, the development of new reactions of difluoroenol silyl ethers is still highly demanding.
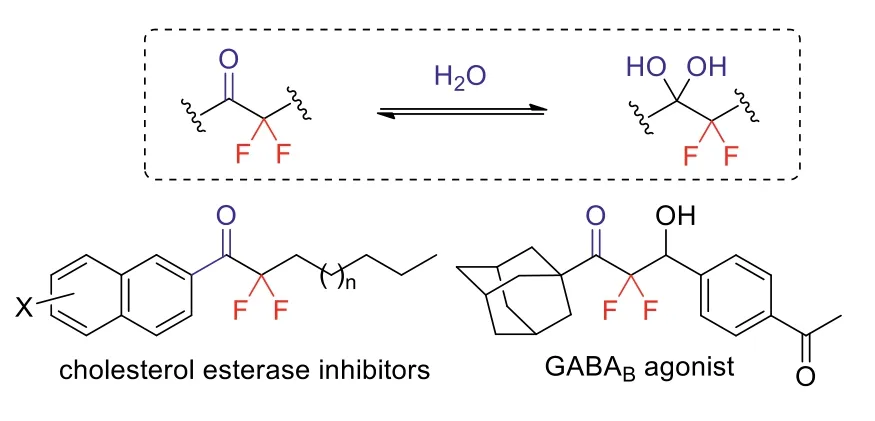
Fig.1.Representative bioactive α,α-difluoroketones.
Alkylnitriles play an important role in organic and medicinal chemistry [44–46].The preparation of alkylnitriles has attracted considerable attention [47–49].Recent years have witnessed a renaissance of ring-opening of cyclobutanone oxime esters for the direct installation of diverse cyanoalkyl moieties into organic compounds under transition-metal [50–58] or photoredox [59–61] catalyzed conditions.Inspired by these achievements, herein we disclose an iron-catalyzed coupling of difluoroenol silyl ethers and cyclobutanone oxime esters to access novelgem-difluoromethylenated ketonitriles (Scheme 1e).
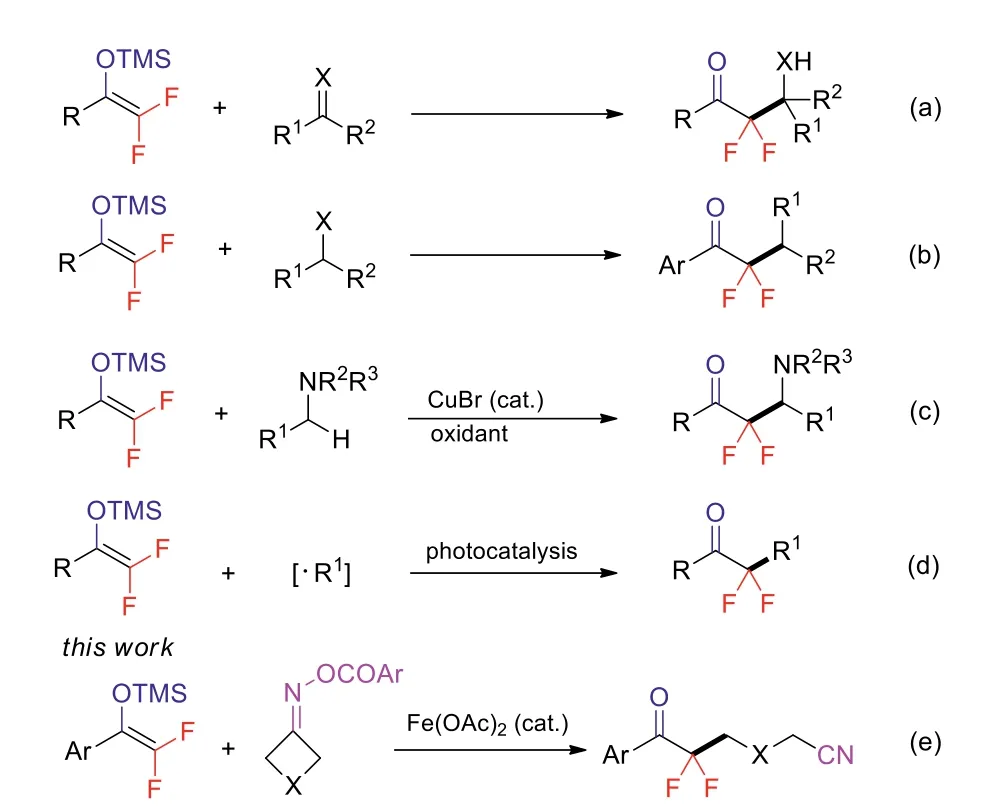
Scheme 1.Reactions with difluoroenol silyl ethers.

Table 1 Optimization of reaction conditions.a
Initially, difluoroenol silyl ether 1a and cyclobutanone oxime ester 2a were chosen as the model substrates to optimize the reaction conditions (Table 1).To our delight, the common metal catalysts, such as Cu(OTf)2, Ni(OAc)2, and Fe(OTf)2, could promote this coupling reaction (entries 1–3).Among them, Fe(OTf)2was optimal, affording the desired product 3aa in 74% yield.Then, other iron catalysts including Fe(acac)2, Fe(OAc)2, and Fe(OTf)3were tested (entries 4–6).The yield of 3aa was improved to 89% with Fe(OAc)2as the catalyst.However, further screening of the solvent(entries 7–10), reaction temperature (entries 11 and 12), and amount of catalyst (entries 13 and 14) failed to give better results.
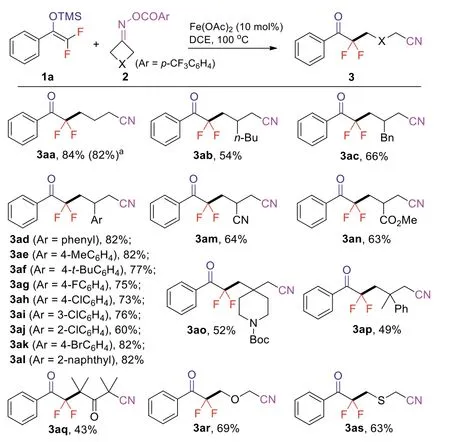
Scheme 2.Scope of cyclobutanone oxime esters.Reaction conditions: 1a(0.4 mmol), 2 (0.2 mmol), Fe(OAc)2 (0.02 mmol), DCE (2.0 mL), N2, 100 °C, 12 h,isolated yields.a Reaction was performed in 1.0 mmol.
With the optimized reaction conditions in hand, we first examined the scope of cyclobutanone oxime esters (Scheme 2).A variety of cyclobutanone oxime esters 2a-2s reacted efficiently with 1a to provide the correspondinggem-difluoromethylenated ketonitriles 3aa-3as in moderate to good yields.3-Substituted oxime esters bearing alkyl (2b), benzyl (2c), aryl (2d-2l), nitrile (2m) and ester (2n) participated in this reaction smoothly.The synthetically useful aryl halides (2g-2k) were well tolerated.It should be noted that the 3,3-disubstituted cyclobutanone oxime ester (2o, 2p) did not exhibit obvious steric effect.On the other hand, sterically hindered substrate 2q deliver the desired product 3aq in lower yield.Oxetan-3-one and thietan-3-one derived oxime esters 2r and 2s were also suitable substrates.Notably, this reaction could be easily scaled up to 1.0 mmol with a slight decrease in yield.
Next, the generality of difluoroenol silyl ethers was investigated.As shown in Scheme 3, aromatic difluoroenol silyl ethers (1b-1g)bearing both electron-donating and electron-withdrawing groups were smoothly converted to the cyanoalkylated products.Naphthyl difluoroenol silyl ether (1h) was found to be the suitable reactant.The cyanoalkylation of heteroaryl difluoroenol silyl ether (1i) was also successful, affording products 3ia and 3id in moderate yields,respectively.However, alkyl difluoroenol silyl ethers were not compatible with the reaction conditions.The structure of product 3dr was confirmed by X-ray crystallographic analysis (for details, see Supporting information).
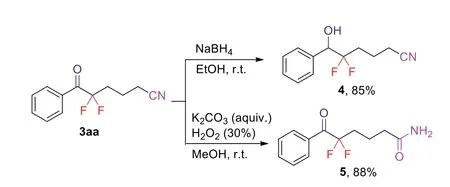
Subsequently, the derivatization of product 3aa was performed to probe the synthetic utility of this protocol (Scheme 4).Reduction of 3aa with NaBH4in EtOH afforded the corresponding alcohol 4 in 85% yield.Furthermore, treatment of 3aa with K2CO3and H2O2in MeOH furnished the amide derivative 5 in 88% yield.
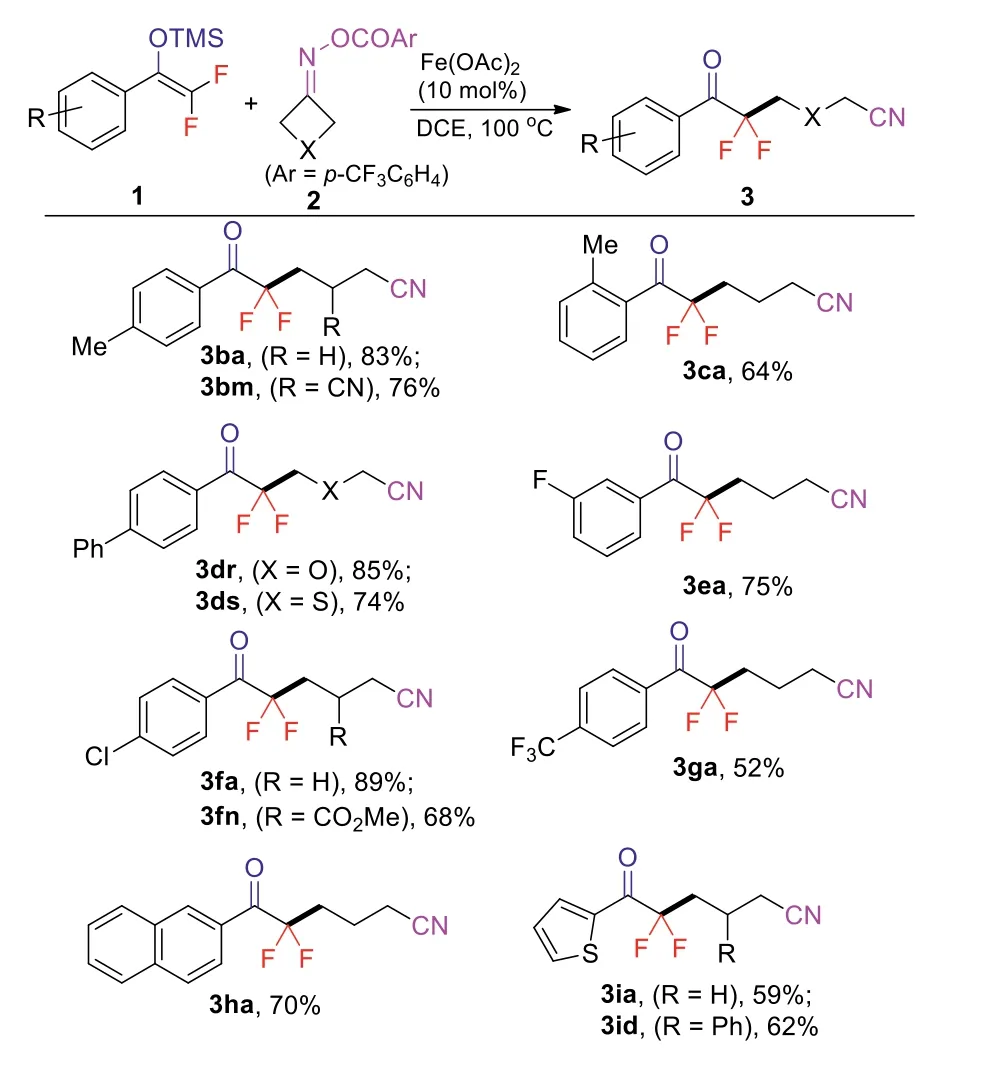
Scheme 3.Scope of difluoroenol silyl ethers.Reaction conditions: 1 (0.4 mmol), 2(0.2 mmol), Fe(OAc)2 (0.02 mmol), DCE (2.0 mL), N2, 100 °C, 12 h, isolated yields.
Scheme 4.Transformation of 3aa.
Scheme 5.Proposed reaction mechanism.
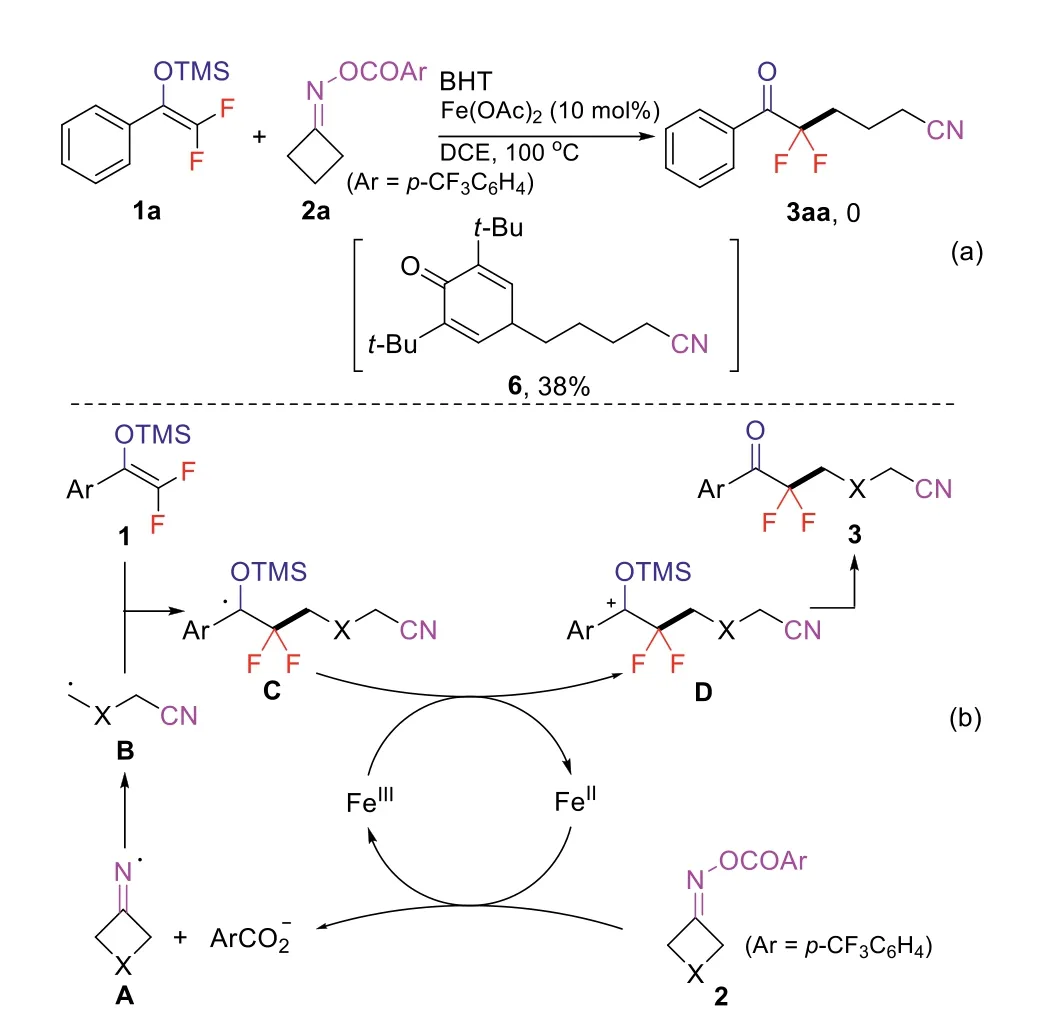
To gain insight into the reaction mechanism, a radical scavenger, 2,6-di-tert-butyl-4-methylphenol (BHT), was added to the reaction mixture.The target product 3aa was not observed; instead, a cyanoalkyl-BHT adduct 6 was formed in 38% yield(Scheme 5a).This result indicated that a cyanoalkyl radical species was probably involved in this reaction.On the basis of this result and previous studies [40–43,50–58], a plausible mechanism was proposed for this iron-catalyzed reaction (Scheme 5b).First, singleelectron transfer (SET) from Fe(II) catalyst to cyclobutanone oxime esters 2 gives iminyl radical A and an Fe(III) species.Then, iminyl radical A undergoes C-C bond cleavagevia β-elimination, leading toγ-cyanoalkyl radical B, which is subsequently added to difluoroenol silyl ethers 1 to giveβ,β-difluoroalkyl radical C.This radical intermediate C would be oxidized by Fe(III) species to the corresponding cation intermediate D.Finally, desilylation of intermediate D furnishes the desired products 3.
In summary, we have developed an iron-catalyzed coupling of readily available difluoroenol silyl ethers and cyclobutanone oxime esters to access novelgem-difluoromethylenated ketonitriles.This reaction proceeds under mild conditions with excellent functional group tolerance.Due to increasing importance of fluorine-containing compounds and synthetic utility of carbonyl and nitrile groups, this protocol might find applications in drug discovery and material science.
Declaration of competing interest
The authors report no declarations of interest.
Acknowledgments
National Natural Science Foundation of China (Nos.21421002,21991211), and the Strategic Priority Research Program of the Chinese Academy of Sciences (No.XDB20000000) are greatly acknowledged for funding this work.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2021.07.030.
 Chinese Chemical Letters2022年2期
Chinese Chemical Letters2022年2期
- Chinese Chemical Letters的其它文章
- Comment on “Acid-induced tunable white light emission based on triphenylamine derivatives”
- Strategies for efficient photothermal therapy at mild temperatures:Progresses and challenges
- Liposome-based delivery of biological drugs
- Macrophage-targeted nanomedicine for chronic diseases immunotherapy
- Advances, opportunities, and challenge for full-color emissive carbon dots
- Fluorine-containing agrochemicals in the last decade and approaches for fluorine incorporation