
Diverse Roles of Immune Cells in Transplant Rejection and Immune Tolerance
2022-06-11 09:04:20XiaojieGanJianGuZhengJuLingLu
Engineering 2022年3期
Xiaojie Gan, Jian Gu, Zheng Ju, Ling Lu*
Hepatobiliary Center, The First Affiliated Hospital of Nanjing Medical University & Research Unit of Liver Transplantation and Transplant Immunology, Chinese Academy of Medical Sciences, Nanjing 210029, China
Keywords:Immune cells Innate immune cells Adaptive immune cells Organ transplant Immune tolerance
ABSTRACT Organ transplant rejection (OTR) is a complex immune reaction involving multiple cells, and it determines graft survival and patient prognosis. At present, most transplant recipients are administered a combination of immunosuppressive and biological agents to protect them from OTR. However,immunosuppressive agents negatively impact the immune system of the patients,causing them to suffer from serious complications, such as chronic infection and malignant tumors. Therefore, a thorough understanding of the mechanisms involved in immune tolerance and immune rejection with regard to organ transplant (OT) is essential for developing better treatment options and improving patient outcomes.This article reviews the role of immune cells in OTR and organ transplant tolerance(OTT),including the novel cell therapies that are currently under clinical trials for transplant recipients.
1. Introduction
An organ transplant (OT) is an effective treatment for patients with end-stage organ failure. Since ancient times, humans have had this idea: If a certain organ of the body has been damaged by disease, can it be replaced like a damaged part in a machine[1,2]? In 1954, Joseph Murray performed the first human kidney transplant between monozygotic twins [3]. Moore et al. [4]described the first orthotopic liver transplant in dogs in 1958,and in 1963,Starzl et al.performed the first human liver transplant[5]. In the same year, D. Hardy conducted the first lung transplant in Jackson, Mississippi [6]. In 1967, Christiaan Barnard performed the world’s first heart transplant at Groote Schuur Hospital in Cape Town, South Africa [7]. Although the technical limits of surgery were overcome during that period, patient mortality due to organ rejection remained high. It was not until the development of cyclosporine, a drug inhibiting the body’s attack on foreign grafts,that OT became a routine treatment for end-stage organ failure patients in the late 1970s. However, transplant tolerance or rejection by the immune system determines graft survival and patient prognosis.
Organ transplant rejection (OTR) is an immunological response to foreign tissue involving various innate and adaptive immune cells. As immunosuppressants are continuously being developed,short-term graft survival has achieved great success, and a oneyear graft survival rate is greater than 80%. However, such immunosuppression strategies do not promote long-term (tenyear) graft survival [8,9]. The general lifespan of a transplanted organ does not exceed 15 years, and in the case of a single lung transplant, it is approximately six years [10]. Therefore, a better comprehension of the mechanisms that determine tolerance or rejection of OTs is essential to develop better immunosuppressive strategies and improve patient prognosis.
This article provides an overview of the role of immune cells in inducing OTR and organ transplant tolerance (OTT), including novel cell therapies that are currently under clinical trials for transplant recipients (Tables 1 and 2 and Fig. 1).
2. Innate immune cells
2.1. Monocytes/macrophages
Macrophages consist of tissue-resident macrophages, and monocyte-derived macrophages recruited from blood vessels and play an essential role in innate immune responses. Macrophages can change their phenotype and function according to the tissuemicroenvironment to attack transplanted organs or prolong graft survival through various suppressive mechanisms [11]. Studies have shown that macrophages activated by interferon (IFN)-γ,lipopolysaccharides(LPS),tumor necrosis factor(TNF)-α,and granulocyte–macrophage colony-stimulating factor(GM-CSF)differentiate into type 1 macrophages(M1),and macrophages stimulated by interleukin (IL)-4 and IL-13 differentiate into type 2 macrophages(M2)[12].Despite substantial research,the diversity and complexity of tissue-specific macrophages in vivo are constantly being revealed, and there are no widely accepted classifications.
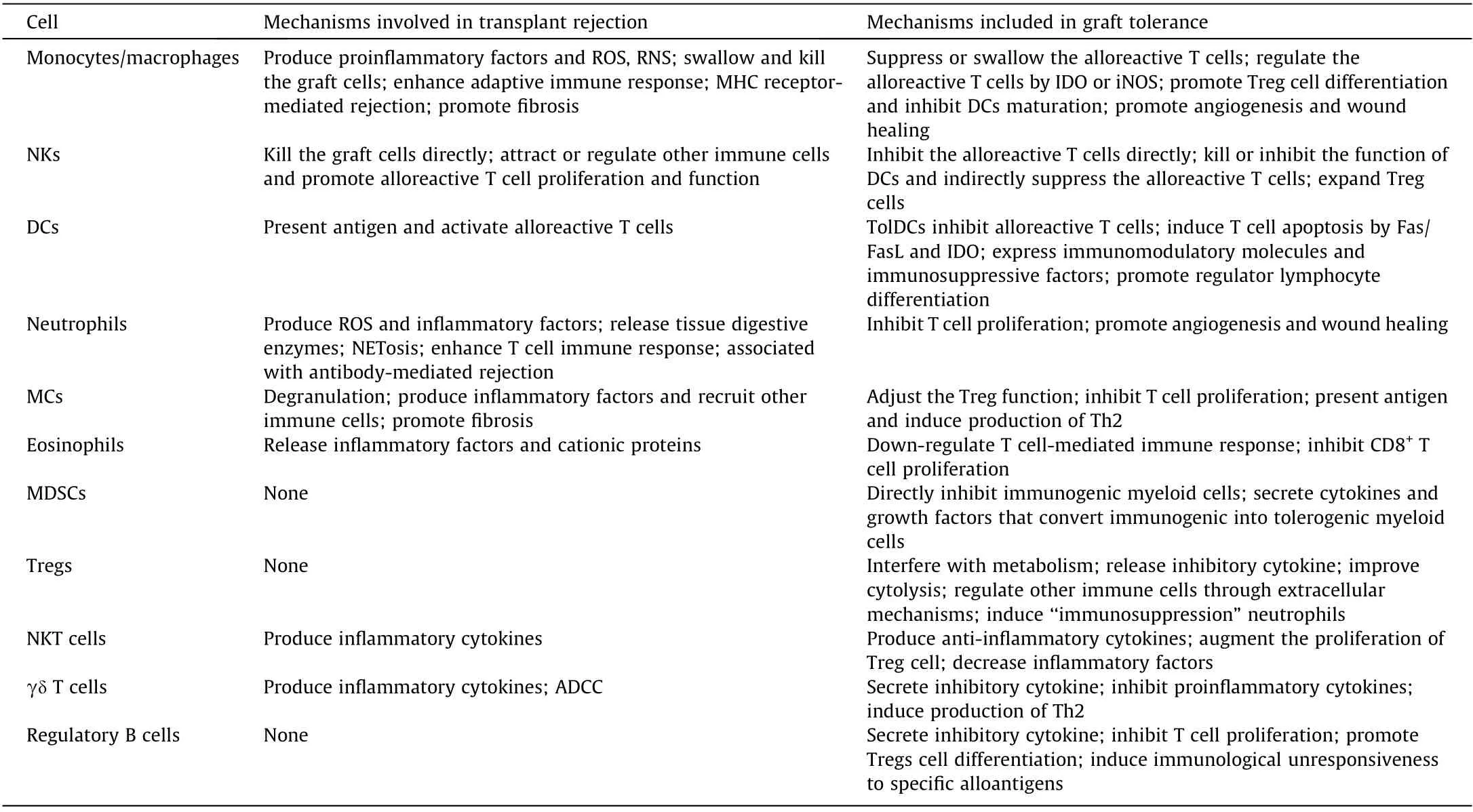
Table 1 The role and mechanism of immune cells in the immune response to transplantation.
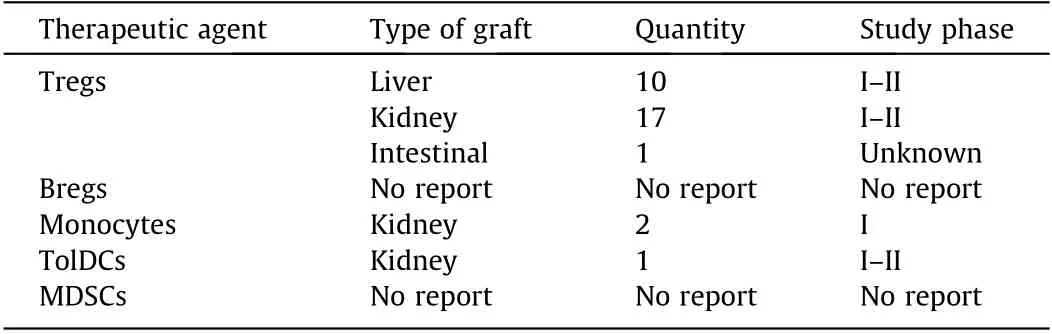
Table 2 Clinical trials of regulatory cell-based therapies in solid organ transplantation(resource from ClinicalTrials.gov).
M1, also known as classically activated macrophages, secrete pro-inflammatory cytokines such as IL-1, IL-6, TNF-α, IL-23, and highly express inducible nitric oxide synthase (iNOS). M1 participate in the immune response to bacterial, fungal, and viral infections; however, their sustained activation inflicts damage on the tissue. In contrast, M2 express platelet derived growth factor(PDGF), insulin-like growth factor-1 (IGF-1), vascular endothelial growth factor (VEGF)-α, anti-inflammatory cytokines, and chemokines, promoting wound healing, angiogenesis, phagocytosis, fibrosis, and the resolution of inflammation.
An increasing number of studies have shown that macrophages are essential in acute OTR. One full-gene transcriptome analysis indicated that the pro-inflammatory macrophage-associated-3 gene was up-regulated during acute graft rejection in a biopsy study. Its expression was positively correlated with the severity of subclinical graft injury [13]. During the early stage of OT ischemia-reperfusion, recipients’ macrophages rapidly infiltrate the graft site, producing many pro-inflammatory cytokines (such as IL-1,IL-12,IL-18,IL-6,IL-23,TNF-α, and IFN-γ),which damages grafts [14]. Additionally, macrophages can also promote acute rejection of transplants by producing reactive oxygen species(ROS) and reactive nitrogen species (RNS) [15,16]. The interaction between RNS and ROS promotes cytotoxic peroxynitrite production and causes peroxidation of the cell lipid membrane.Moreover,macrophages mediate acute OTR by activating adaptive immune responses. As an antigen-presenting cell (APC), both donor- and recipient-derived macrophages can present antigens and activate T cells through co-stimulatory signals on the cell surface to release pro-inflammatory cytokines, resulting in acute rejection.
Similarly,macrophages are associated with chronic rejection of OT. Chronic rejection of allografts is characterized by interstitial infiltration of macrophages and T cells, and increased macrophage infiltration in the allograft is positively associated with graft failure[17,18].M2 accelerate chronic graft rejection by promoting smooth muscle cell proliferation and interstitial fibrosis.Recipient biopsies indicate that M2 dominate the grafts within chronic rejection,and their number is positively correlated with the degree of fibrosis[19]. Conversely, inhibition of infiltrating M2 by oxidized adenosine triphosphate (ATP), whose receptor P2x7r is preferentially expressed in M2,reduces the degree of graft vascular disease and fibrosis and prolongs the survival of heart grafts [20]. Macrophages also promote interstitial fibrosis of the grafts by transforming into myofibroblasts,characterized by the co-expression marker of macrophages (CD68) and myofibroblasts (α-smooth muscle actin (α-SMA)).
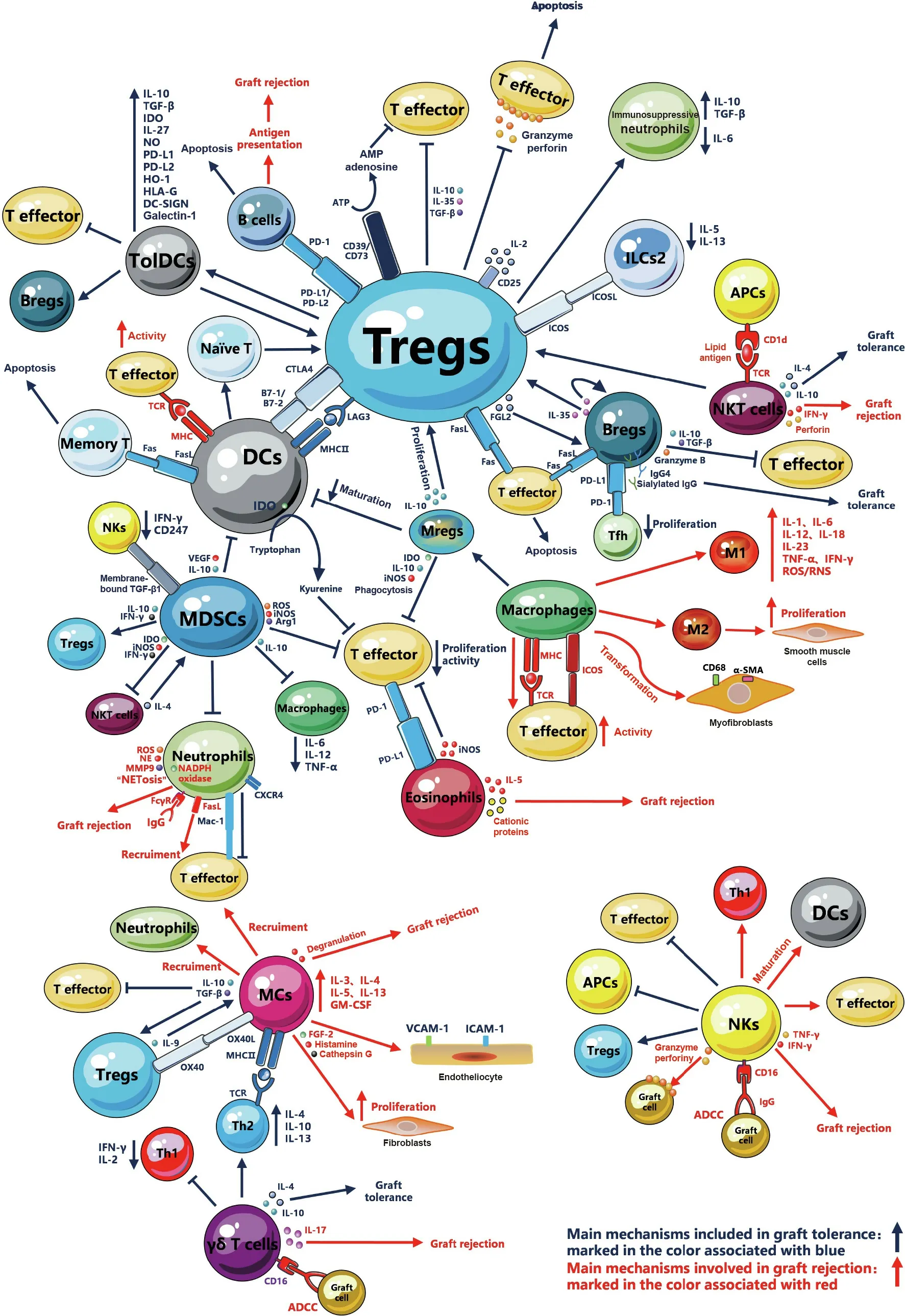
Fig.1. Schematic diagram of the interaction among the immune cells in transplant immune response.The main mechanisms involved in graft tolerance are marked in blue and those involved in graft rejection are marked in red. ICOSL: inducible T cell co-stimulator ligand; FGF: fibroblast growth factor; FcγR: Fc gamma receptor.
Although macrophages promote OTR through various mechanisms,recent studies have shown that the adoptive transfer of regulatory macrophages(Mregs)can induce immune tolerance of the transplanted organs. Mregs are macrophages activated by macrophage colony stimulating factor (M-CSF) and IFN-γ and constitute a subpopulation of macrophages with unique phenotypes and inhibit alloreactive T cell proliferation and function. Mregs secrete IL-10 via fucosylated ligand-mediated dendritic cell-specific intercellular cell adhesion molecule-grabbing non-integrin(DC-SIGN) and Toll-like receptor 4(TLR4)pathways to inhibit CD8+T cell immune activity and promote CD4+forkhead box P3 (Foxp3)+regulatory T cells (Tregs) amplification [21]. Studies show that mouse Mregs inhibits T cell activity by iNOS and eliminates alloreactive T cells by phagocytosis in vitro[22].Human Mregs specifically expressing DHRS9 inhibit T cell proliferation by activation of indoleamine 2,3-dioxygenase (IDO) via IFN-γ and contact-dependent mechanisms[23,24]. Moreover, human Mregs induce T cell immunoreceptor with immunoglobulin and ITIM domain (TIGIT)+Foxp3+iTregs differentiation and inhibit dendritic cell maturation to promote OTT[25].
2.2. Natural killer cells
Natural killer cells (NKs) are important components of the innate immune system,accounting for 10%–15%of peripheral lymphocytes, and play a key role in immune surveillance, including tumor surveillance, defense against viruses, and allograft immunoreactivity [26,27]. Human NKs are characterized as CD3--CD56+CD335(NKp46)+lymphocytes and can be further divided into two subgroups based on the expression level of CD56:low expression (CD56dim) and high expression (CD56bright) [28].
CD56dimNKs are mainly found in peripheral blood and express high levels of CD16 (FcγRIII) and terminal differentiation marker CD57. They release perforin and granzyme to kill target cells directly. CD56brightNKs are mainly found in secondary lymphoid organs,express low amounts of CD16,mediate immune responses by secreting pro-inflammatory cytokines (e.g., IFN-γ and TNF-α),and induce apoptosis by expressing membrane TNF family molecules factor associated suicide ligand (FasL), TNF-related apoptosis-inducing ligand (TRAIL), and transmembrane TNF(mTNF), which bind to target cell membrane ligands [29,30].
Research has shown that NKs are not involved in solid OTR, as severe combined immunodeficiency (SCID) or recombinationactivating protein (Rag)-/-mice (lacking T and B cells but have functional NKs) are tolerant to allografts [31]. However, as research continues, experts are increasingly aware that NKs also participate in the transplant rejection of solid organs[32].The balance of activating and inhibitory signals determines NKs function.Early post-transplant, NKs infiltrate the allografts [33] and can be activated by allograft antigens or cytokines (e.g., IL-12, IL-2, and IFN-γ) secreted by dendritic cells (DCs) and T cells [34]. Activated NKs can kill alloreactive target cells either directly or by releasing cytokines. Studies have shown that NKs activated by IL-15 contributed to skin allograft rejection in Rag-/-mice [35]. In a study of gene expression profiling in human kidney biopsy tissue, high levels of NK transcripts were found, suggesting that NKs have a unique role in this rejected kidney [36,37]. The pathology of renal transplant rejection is divided into two categories:T cell-mediated rejection(TCMR)and antibody-mediated rejection(AMR)[38].The secretion of pro-inflammatory cytokines (e.g., IFN-γ and TNF-α)remains the primary cause of TCMR mediated by NKs. These cytokines can:
(1) Up-regulate the chemokines (e.g., chemokine (C-X-C motif)ligand 9 (CXCL9)) secreted by NKs to recruit alloreactive T cells and promote T helper type 1 cell (Th1) response [39–41];
(2) Up-regulate the expression of major histocompatibility complex (MHC)-II and co-stimulatory molecules on DCs to promote DCs maturation [42];
(3) Promote Th1 differentiation and transplant rejection[43];
(4) Up-regulate human leucocyte antigen (HLA) alloantigen on donor tissue grafts, marking them for destruction by NKs [44].
Recent transcriptomic evidence suggests that NKs activation is triggered by the surface antigen CD16 (IgG Fc) receptor in AMR.The donor-specific antibody (DSA) binding to allograft endothelial cells interacts with CD16 on NKs to induce antibody-dependent cell-mediated cytotoxicity (ADCC) on allografts [45].
Interestingly, NKs also induce tolerance in allografts, which can occur by inhibiting alloreactive T cells and APCs functions.NKs can specifically inhibit donor alloreactive T cells in the mouse graft-versus-host disease (GVHD) model to promote immune tolerance [46]. Additionally, NKs can directly kill donor-derived DCs by releasing perforin, granzymes, or other mechanisms, thereby suppressing immune responses and promoting the formation of a tolerogenic environment [47]. In a mouse skin graft model, recipient NKs kill donor APCs, thereby inhibiting alloreactive T cell proliferation and promoting tolerance to allogeneic skin [48]. Immunoregulatory NKs (NKreg) have also been shown to inhibit antigen-specific T cells in vitro cell culture [49]. Trojan et al. [50] showed that renal transplant recipients who survived for more than one and a half year might have NKregin their bodies. The function of NKreg, including secreting IFN-γ and ADCC, is also weakened in a similar way to NKregin the uterus to protect pregnancy [51]. Yu et al. [52] demonstrated that alloreactive NKs promote tolerance to hemizygous hematopoietic stem cell transplant by amplifying recipientderived CD4+CD25+Tregs.
2.3. Dendritic cells
DCs are regarded as the most important APCs because they can initiate an immune response by activating T and B cells, functioning as the bridge between innate and adaptive immune systems.DCs are derived from hematopoietic stem cells in the bone marrow and have complex heterogeneity. Human DCs can be divided into classical/conventional DCs (cDCs), including myeloid DCs (mDCs),lymphoid DCs, and plasmacytoid DCs (pDCs), which are capable of secreting large amounts of type I interferons [53]. Functionally,they can be divided into ‘‘mature” DCs and ‘‘immature” DCs(imDCs).
After an OT, donor DCs migrate to the recipient’s secondary lymphoid organs and induce alloreactive naive T cells to differentiate into effector T cells, which in turn migrate into the graft and mediate rejection.Recipient T cells recognize alloreactive antigens directly, semi-directly, and indirectly. Direct recognition occurs when recipient T cells directly recognize the alloreactive MHC molecules on the surface of donor DCs,which usually triggers acute rejection. Semi-direct recognition occurs when recipient T cells indirectly identify the donor antigen peptide presented by the MHC on the recipient DC surface and directly identify the donor antigen peptide–donor MHC molecule complex that is transferred onto the surface of the recipient DCs[54,55].Indirect recognition means that recipient T cells recognize donor antigen peptides processed and presented by recipient DCs,which is a factor in late stage rejection and chronic rejection.
Studies have shown that ‘‘immature” tolerogenic DCs (TolDCs)can promote tolerance to alloreactive antigens [56]. It is now understood that TolDCs can promote allogeneic graft tolerance via the following mechanisms:
(1) Expression of low levels of MHC class II molecules and costimulatory molecules to induce T cell anergy and clonal deletion;
(2) Activation of the factor associated suicide/factor associated suicide ligand (Fas/FasL) pathway and IDO expression to initiate apoptosis in naive and memory T cells;
(3) Amplification or induction of regulatory lymphocytes (e.g.,CD4+CD25hiFoxp3+Tregs, lymphocyte-activation gene (LAG)-3+-CD49b+CD25+Foxp3+/-Tregs (Tr-1), CD8+Tregs, regulatory B cells(Bregs), and T cell receptor (TCR) αβ+CD3+CD4-CD8-NKRP1-double-negative T cells (DNT cells)) to induce immune tolerance;
(4)Production of immunosuppressive factors(e.g.,IL-10,transforming growth factor (TGF)-β, IDO, IL-27, and nitric oxide (NO))and expression of immunoregulatory molecules(e.g.,programmed death ligand-1 (PD-L1), PD-L2, heme oxygenase (HO)-1, human leucocyte antigen (HLA)-G, TNF-related apoptosis-inducing ligands, galectin-1, and DC-SIGN) to promote central and peripheral immune tolerance [57].
Mouse experiments have shown that injection of donor-derived imDCs seven days before the allogeneic heart transplant can significantly prolong graft survival[58].Additionally,injection of donorderived DCs can prevent graft rejection of the skin and GVHD[59,60]. pDCs can also promote transplant immune tolerance[61]. In a mouse model, pDCs presenting alloantigen migrate to draining lymph nodes and induce Treg production. Studies have shown that in liver transplant patients free of immunosuppressive agents, the expression of PD-L1 and CD86 by pDCs was positively correlated with the number of CD4+CD25+Foxp3+Tregs [62].
2.4. Neutrophils
Neutrophils are small phagocytic cells derived from bone marrow stem cells and account for 50%–70%of peripheral blood leukocytes. They express IgG Fc receptors and play a pivotal role in phagocytosing and destroying foreign matter via complementmediated or antibody-dependent pathways. After an OT, neutrophils are first responders present in the graft and express pattern recognition receptors (PRRs) binding to damage associated molecular patterns (DAMPs) released by necrotic cells in extracellular matrix(ECM)to induce ROS and hydrolase production,which exacerbates graft ischemia-reperfusion injury (IRI) [63].
In the IRI phase of grafts,neutrophils destroy grafts and exacerbate rejection using the following mechanisms:
(1)Production of superoxide through the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase system to promote macromolecular peroxidation and irreversible cellular damage of the graft tissue [64,65];
(2) Release of tissue digestive enzymes such as metalloproteinase (MMP)-9 and neutrophil elastase (NE) to break the steady barrier that promotes graft function [66];
(3) Neutrophil extracellular trap formation (‘‘NETosis”) to promote inflammation in the graft [67].
It has been reported that in the patient of primary lung graft dysfunction and the mouse model of liver IRI mediated by a lung transplant, researchers have used deoxyribonuclease (DNAse) to solubilize NETs, which reduces acute inflammatory responses and significantly improves graft function [68,69].
The adaptive immune response initiated by neutrophils is a primary cause of graft rejection. Neutrophils can induce chemokine(C–C motif) ligand (CCL)1, CCL2, and CCL5 expression in T cells via FasL/perforin-mediated activity to recruit activated CD8+T cells[70]. Researchers found acute rejection can be alleviated in the mouse skin graft model by depleting neutrophils to attenuate the recruitment of alloreactive memory CD8+T cells [71]. Moreover,in a mouse orthotopic lung transplant model,the depletion of neutrophils promotes immunosuppression-mediated tolerance and reduces the production of IL-12 by graft APCs and alloreactive immune responses by Th1 cells [72].
Contrary to TCMR, clinical pathology suggests that increasing neutrophils in the graft is associated with AMR. In mouse cardiac and lung transplant models, AMR induces neutrophil infiltration which destroys transplants [73]. Studies have shown that increasing neutrophils infiltration is also related to chronic graft rejection[74]. In chronic rejection, Th17 cells attract more neutrophils to accumulate in local sites by secreting IL-17 and then mediate graft rejection by the mechanisms mentioned above.Further,it has been reported that in lung transplant recipients, IL-17-mediated neutrophil infiltration increases the risk of chronic rejection [75].
Some studies suggest that neutrophils are not conducive to the formation of graft immune tolerance. However, others indicate that regulatory subpopulations in neutrophils are equipped with inducible anti-inflammatory properties, which can protect allografts and promote tolerance. Pillay et al. [76] have shown that CD16bbrightCD62loneutrophil subpopulations in patients with an acute injury can bind to T cells via macrophage-1 antigen (Mac-1) and release hydrogen peroxide, thereby inhibiting T cell proliferation. Moreover, neutrophils can form dense clusters around the necrotic tissue through their integrin receptors,isolating them from healthy tissues and promote healing and tissue repair to inhibit graft inflammation [77].
Christoffersson et al. [78] discovered a neutrophil subpopulation marked with CD11b+Gr-1+CXCR4hi, which can be recruited to the avascular islet grafts of mice model in a VEGF-Adependent manner and is helpful to reconstitute islet perfusion.Additionally, neutrophil subpopulations characterized by CD49+-VEGFR1hiCXCR4hiwith similar pro-angiogenic functions were also found in humans [79].
2.5. Mast cells
Mast cells (MCs) are a type of granular immune cell which differentiate from CD34+/CD117+multipotent progenitor cells in the bone marrow [80]. Studies have shown that MCs regulate innate and adaptive immune responses and play a key role in forming immune tolerance and rejection in allogeneic OT [81].
The main mechanisms of MCs participating in OTR are described below.
(1) Degranulation: Studies have shown that the use of MC stabilizer Cromolyn to inhibit MCs degranulation can improve bronchiolitis obliterans and pulmonary fibrosis in allografts and prevent allograft lung transplant rejection [82];
(2) Cytokine secretion: By secreting GM-CSF, TNF-α, IL-3, IL-4,IL-5, and IL-13 up-regulates the vascular cell adhesion molecule(VCAM)-1 and intercellular cell adhesion molecule (ICAM)-1 of the endothelial cells and recruits neutrophils and T cells to graft[83];
(3) Fibroblasts activation: In chronic rejection of kidney, lung,and heart transplants, MCs release fibrogenic mediators (e.g., histamine, fibroblast growth factor-2, TGF-β, heparin, cathepsin G,and chymotrypsin) to activate fibroblasts, promote collagen synthesis, and ultimately induce graft fibrosis [83].
MCs play a key role in Tregs-mediated peripheral immune tolerance. Tregs promote the migration of MCs to grafts by secreting MCs growth factor IL-9 [84]. Tregs stabilize MCs and inhibit IgEmediated degranulation by interacting with MCs via tumor necrosis factor receptor superfamily member 4(OX40)/OX40 ligand[85].Conversely,MCs can release TGF-β,IL-10,and specific proteases to inhibit T cell proliferation and promote Tregs production [86].Moreover, MCs expresses the MHC-II molecule, which can present antigens to T cells, induces the production of Th2 cytokines (e.g.,IL-4, IL-10, and IL-13), inhibits IFN-γ production, and participates in the transformation of immature T cells to Th2 cells, which is conducive to tolerance.
2.6. Eosinophils
Eosinophils, named for their rich eosinophilic granules, are derived from bone marrow stem cells and undergo phagocytosis.They are mainly involved in rapid-onset hypersensitivity, antiparasitic and viral infections. Studies have shown that eosinophils also mediate allograft rejection. Eosinophils cause tissue damage and rejection by expressing cationic granule protein and cytokines such as IL-5, and attenuating IL-5 can reverse graft rejection [87].Increased eosinophils counts in peripheral blood and graft biopsy tissues have been associated with acute rejection in kidney, heart,and lung transplants[88–91].It has been reported that the number of eosinophils is increased in bronchoalveolar lavage and blood of lung transplant recipients diagnosed with restrictive chronic lung allograft dysfunction (rCLAD), which was also related to the low survival rate of transplants [92]. Eosinophils participate in liver regeneration and play an essential role in predicting acute liver rejection [93,94]. The Royal Free hospital regards the increase of eosinophils in graft biopsy as a key component in diagnosing acute rejection [95,96].
Recent studies have shown that eosinophils can also promote tolerance in the lung transplant mice model [97]. Onyema et al.[97] demonstrated that eosinophils could down-regulate T cellmediated immune response, and this down-regulation depended on synapse formation mediated by PD-L1/ programmed death-1(PD-1) between eosinophils and T cells. Th1-polarized eosinophils can interfere with TCR/CD3 subunit binding and signal transduction in an iNOS-dependent manner, thus inhibiting the proliferation of CD8+T cells in the graft [98].
2.7. Myeloid-derived suppressive cells
Myeloid-derived suppressive cells (MDSCs) are immature,highly heterogeneous cells developing from bone marrow. MDSCs differentiate into macrophages, DCs, and granulocytes depending on their microenvironment in vivo, and have immunosuppressive properties [99]. Most human MDSCs express CD11b, CD33, CD34,and MHC class II molecules, while CD11b and GR1 are expressed in mice. Based on their morphology, MDSCs can be divided into granulocytic MDSCs (G-MDSCs) and monocytic MDSCs (MMDSCs), and can be further subdivided according to Ly6C and Ly6G expression. However, the surface marker of MDSCs remains controversial [100]. MDSCs are thought to exert their induction of immune tolerance using the following mechanisms:
(1) Direct inhibition of immunogenic bone marrow cells (e.g.,macrophages, neutrophils, and DCs);
(2) Secretion of cytokines and growth factors (e.g., iNOS, arginase (Arg)-1, HO-1, ROS, IDO, IL-10, and TGF-β1) to transform immunogenic bone marrow cells into tolerogenic cells [101].
In 2008,Dugast et al.[102]first discovered the accumulation of CD11b+CD80/86+Sirpα+myeloid cells in blood and grafts of rat allogeneic kidney transplant tolerance model and defined them as MDSCs. MDSCs can inhibit effector T cells proliferation, induce iNOS-dependent apoptosis, and induce Tregs amplification[103,104]. A transplant tolerance mechanism of MDSCs was also identified in mice models of corneal, islet, skin, and heart transplants [105–107]. Hock et al. [108] observed an increase in the number of MDSCs in kidney transplant recipients,especially those with renal squamous cell carcinoma who underwent a kidney transplant,and found that recipients with high numbers of MDSCs survived longer than recipients with low MDSCs counts[109].In a prospective cohort study of 36 intestinal transplant recipients,researchers identified three types of MDSCs,all of which were able to inhibit CD4+and CD8+T cell proliferation and IFN-γ production,and promote the survival of intestinal transplants [110]. Although the above studies indicate MDSCs promote tolerance of transplants, the exact mechanisms require further research.
3. Adaptive immune cells
3.1. Regulatory T cells (including CD4+ T cells, CD8+ T cells, and CD4-CD8- T cells)
Studies have shown that many T cell subsets play a role in OTT,including CD4+T cells,CD8+T cells,CD4-CD8-T cells,natural killer T (NKT) cells, and γδ T cells.
3.1.1. CD4+regulatory T cells
CD4+Tregs are the most intensively investigated Treg subset.Tregs can inhibit allograft rejection and GVHD responses [111].Tregs from the thymus (tTregs), also called natural Tregs (nTregs),migrate to the periphery and inhibit autologous antigens immune responsivity. Tregs phenotypes are heterogeneous, but Foxp3 is a characteristic marker of CD4+Tregs.Antigens can also induce Tregs to express Foxp3 within the periphery microenvironment of tolerance, called adaptive or induced Tregs (iTregs). Both tTregs and iTregs recognize and respond to alloantigens. However, studies have shown that Tregs, persistently in response to alloantigens,play a more important role in OTT [112].
Although there is growing evidence that alloreactive T cells participate in allograft destruction and cause irreversible damage to the transplanted tissue, Tregs can inhibit T cell function and protect the graft from damage. Therefore, regulating the balance between alloreactive T cells and Tregs is important to preserve the donor graft. Before and after OT, adoptive transfer of Tregs can increase the number of Tregs in recipients and induce immune tolerance.Tregs can inhibit the activity of immune cells and participate in inducing tolerance to the OT through a variety of mechanisms, including:
(1) Secretion of anti-inflammatory cytokines (e.g., IL-10, TGF-β,and IL-35) to inhibit the proliferation of effector T cell;
(2)Release of granzyme and perforin to promote cell apoptosis;
(3) Expression of CD39/CD73 to inhibit the proliferation of effector T cells by depleting ATP in the extracellular microenvironment through the production of adenosine and AMP(immunosuppressive molecules);
(4) Overexpression of CD25 and uptake of more IL-2 in the microenvironment to starve IL-2-dependent cells (e.g., effector T cells and NKs);
(5) Interaction with B cells via PD-L1/PD-1 to inhibit autoreactive B cells in an antigen-specific manner, or release granzyme B and perforin to kill B cells;
(6) Expression of cytotoxic T lymphocyte-associated antigen 4(CTLA4)to inhibit the antigen presentation of DCs and T cell proliferation and promote the induction of Tregs by inducing DCs to produce IDO;
(7) Expression of lymphocyte activation gene (LAG)-3, a molecule with higher affinity than CD4 binding to MHC-II, to decrease antigen presenting ability of DCs and inhibit T cell activation;
(8) Induction of monocytes to differentiate into M2;
(9) Induction of immunosuppressive phenotype neutrophils to resolve inflammation in the local environment;
(10) Expression of inducible T cell co-stimulator (ICOS) to decrease the secretion of IL-5 and IL-13 by intrinsic lymphocyte(ILCs) 2 [113–115].
It has been reported that blocking the activity of CTLA4 or IL-10 in animal transplant models prevents Tregs-mediated immune regulation[116],and many animal models’acute and chronic allograft rejection can be controlled via Tregs adoptive transfer [117].
3.1.2. CD8+regulatory T cells
Like CD4+Tregs, the phenotype of CD8+Tregs is also heterogeneous,and even CD8+Foxp3-Tregs exist[118].Studies have shown that CD8+CD28-Tregs can inhibit immunoreactivity against transplanted kidneys in renal transplant recipients taking alemtuzumab as their induction therapy [119]. It has also been reported that a group of CD8+Tregs can produce IL-10 to induce tolerance in patients receiving an allogeneic kidney transplant. In one study,this group of CD8+Tregs were differentiated from immature CD8+T cells and exerted their action through an IL-10 dependent mechanism [120]. Recently, CD8+Tregs expressing IL-2 receptor β chain(CD122)were found to inhibit islet and skin allograft rejection in an IL-10 dependent manner and were more effective at OTT than CD4+Tregs [121].
The mechanisms of CD8+Tregs in the modulation of immune tolerance in OT may be involved in multiple pathways:
(1)CD8+Foxp3+Tregs express CTLA4 and promote the formation of transplant tolerance through the same mechanism as CD4+Tregs mentioned above [122];
(2) CD8+CD28-Tregs up-regulate the expression of DCs immunoglobulin-like transcripts 3/4 (ILT3 and ILT4), downregulate co-stimulatory molecules (CD80/CD86) and adhesion molecules (CD54/CD58) to make DCs tolerogenic [123];
(3) CD8+CD45RClowTregs overexpress glucocorticoid-induced tumor necrosis factor receptor-related protein (GITR) and interact with alloreactive APCs to inhibit T cell proliferation [124];
(4) CD8+CD122+Tregs directly recognize activated T cells through MHC-I and then produce IL-10 to inhibit immune response[125];
(5) CD8+CD122+PD-1+Tregs interact with APCs via PD-1/PD-L1 and promote IL-10 production to inhibit T cell activation[126,127];
(6) CD8+Tregs promote graft immune tolerance by secreting cytokines such as IL-34, IFN-γ, and TGF-β [128–131];
(7) CD8+Tregs interact with FcγRIIB receptor by secreting fibrinogen-like (FGL) 2 to inhibit DCs maturation, induce B cell apoptosis, and promote Bregs production [132,133];
(8) CD8+Tregs mediate target cell apoptosis by releasing perforin and induce T cell apoptosis by expressing FasL, which interacts with Fas on T cell surface [134];
(9)CD8+Tregs highly express CD122 and CD25,and exhaust IL-2 in the microenvironment to make other IL-2-dependent cells‘‘hungry” (e.g., effector T cells and NKs) [135].
Since MHC-I is expressed in all nucleated cells,the advantage of CD8+Tregs over CD4+Tregs cells lies in the persistence of donor MHC-I presentation.Immune tolerance induced via the direct presentation of MHC-I+graft cells to alloreactive CD8+Tregs is longterm, while direct or indirect presentation by donor APCs to CD4+Tregs is short-term. Studies have shown that in rat heart allograft models, the consequence of donor antigen indirectly presented to CD8+Tregs is even more effective than directly identifying donor cells to inhibit the alloreactive response of effector T cells [136].
Immune regulation mediated by CD4+Tregs is complementary to that of CD8+Tregs.CD4+Tregs mainly inhibit naive effector T cell response,but not memory T cell response,while CD8+Tregs inhibit memory effector T cell response [137]. Additionally, the cytokines produced by these two Tregs can promote the formation of the tolerogenic environment in vivo. TGF-β and IL-10 secreted by CD8+Tregs can promote the expansion of CD4+Tregs, while IL-34 secreted by CD8+CD45RClowTregs can induce the production of Mregs, CD4+Tregs, and CD8+Tregs. It has been reported that in rat heart allograft models,the treatment of IL-34 can induce CD4+-CD25+Tregs and CD8+CD45RClowTregs simultaneously, and transferring these two Tregs to new transplant recipients can produce the tolerogenic effect as well [129].
3.1.3. CD4–CD8–regulatory T cells
CD4-CD8-Tregs (DN Tregs) express CD3 and αβTCR, but not CD4, CD8, or NK1.1. Animal model studies have shown that DN Tregs can prevent CD4+and CD8+T cell-mediated immune response and allograft rejection [138]. DN Tregs can inhibit immune responses in many ways:
(1) DN Tregs can induce T cell apoptosis by the CD95–CD95L pathway;
(2) DN Tregs can express a high level of CTLA4 and downregulate co-stimulatory molecules CD80 and CD86 on DCs to induce apoptosis of DCs;
(3)DN Tregs can induce B cell apoptosis by perforin-dependent pathways [139];
(4) DN Tregs can obtain the whole MHC–antigen complex presented by APCs using trogocytosis and then combine it with CD8+T cells through the MHC–antigen complex to mediate apoptosis via the Fas/FasL pathway [140]. Studies have shown that the adoptive transfer of DN Tregs can prolong graft survival and increase Foxp3+Tregs in the mouse heart allograft models [141].It has also been demonstrated that injection of DN Tregs can effectively inhibit graft rejection in islet, skin, and hematopoietic stem cell transplants [142].
3.1.4. Natural killer T cell
NKT cells are a group of distinct regulatory T lymphocytes simultaneously expressing T cell (TCR and CD3) and NK surface markers(CD56 or NK1.1)[143].NKT cells rapidly secrete cytokines(e.g.,IL-4,IL-10,and IFN-γ)after recognizing CD1d/lipid antigen by TCR. NKT cells can secrete perforin or kill target cells via the Fas/FasL pathway, leading to OTR. NKT cells are also associated with graft tolerance [144]. Ikehara et al. [145] showed that Valpha14+NKT cells promoted allograft immune tolerance in the mouse islet transplant model.In the mouse hematopoietic stem cell transplant model,the adoptive transfer of NKT cells inhibits the development of acute GVHD and IFN-γ, and TNF production [146]. Recent studies have also shown that the adoptive transfer of invariant NKT cells can significantly improve cGVHD by amplifying donor Tregs in a mouse cGVHD model [147].
3.1.5.γδ T cells
γδ T cells are highly conserved lymphocyte subpopulations,accounting for 0.5%–6% of total circulating lymphocytes, 4%–10%of circulating CD3+T cells, and 10%–50% tissue-resident T cells[148,149]. γδ T cells are heterogeneous and can be classified into Vδ2+and Vδ2-γδ T according to the TCR δ chain [150,151]. As a bridge between innate and adaptive immunity, γδ T cells play a significant role in allograft rejection and immune tolerance [152].In a mouse kidney IRI model, early infiltration of γδ T cells after ischemic injury resulted in αβ T cell infiltration and subsequent tubular damage. At the same time, kidney IRI improved with γδ T cell knockout [153]. Studies have shown that if cytomegalovirus(CMV)infection occurs in transplant recipients,leading to the proliferation of CMV-specific Vδ2-γδ T cells,renal allograft injury and acute rejection will be caused by DSA-mediated ADCC activity[154,155].
In a mouse lung transplant model, IL-17+γδ T cells were activated in a TCR-dependent or independent pathway and secreted IL-17, contributing to acute rejection [156,157]. Similarly, in a mouse heart transplant model, γδ T cells producing IL-17 were associated with acute and chronic rejection of the graft, and depletion of γδ T cells reduced serum IL-17 and inflammatory cell infiltration, prolonging graft survival [158,159].
While the adaptable role of γδ T cells in both rejection and tolerance continues to be explored,increasing evidence shows that γδ T cells play a role in immune tolerance. Studies have found that,compared with healthy controls of the same age, the number of γδ T cells was significantly increased, and the ratio of γδ T cells expressing Vδ1 and Vδ2 was altered (Vδ1:Vδ2 >1) in spontaneously tolerant liver transplant recipients [160]. Vδ1 γδT cells are capable of producing IL-10 and promoting Th2 production,while reversing acute rejection of liver transplant associated with elevated Vδ2 γδ T cells[161,162].Before a skin transplant,injecting the recipient with hybridoma cells in the portal vein can induce the expansion of oligoclonal γδ TCR+cells, increasing IL-4 and IL-10 production, inhibiting IL-2 and IFN-γ production, and improve graft survival rate [163]. Additionally, in animal models of small bowel and islet transplant, increased infiltration of γδ T cells improved graft rejection [164,165].
3.2. Regulatory B cells
B cells have multiple immune system functions and can mediate allograft rejection by presenting antigens and producing cytokines and antibodies.However,B cells can also be manipulated to inhibit allograft rejection [166]. Bregs have been identified in humans and mice and are capable of secreting anti-inflammatory cytokines such as IL-10 and IL-35.Bregs have complex heterogeneity with different phenotypes and regulatory functions in humans and mice. Mouse Bregs usually express elevated levels of CD1d,CD5, CD21, CD24, and IgM, while human Bregs express CD19,CD24,and CD38[167].One study of a mouse experimental autoimmune encephalomyelitis model showed that Bregs inhibited inflammatory responses, and the adoptive transfer of LPSactivated Bregs protected non-obese diabetic mice against diabetes[168,169]. The consumption of Bregs in humans promoted psoriasis in multiple sclerosis (MS) patients, exacerbated inflammation,and aggravated ulcerative colitis [170]. Moreover, in OT, TIM-1+Bregs can prolong the survival of mouse allografts. In contrast,TIM-1-/-mice showed defects in IL-10+Bregs and demonstrated accelerated graft rejection; however, after the adoptive transfer of TIM-1+Bregs, graft survival was significantly prolonged[171,172]. The mechanism of Bregs participating in the induction of immune tolerance in OT may be as follows:
(1)Inhibition of the release of pro-inflammatory cytokines(e.g.,IFN-γ and IL-17)from Th1,CTLs,and Th17 via secretion of IL-10 to suppress monocyte activation and DC maturation and induction of Tregs by CD80/CD86;
(2)Inhibition of the activation of Th1,Th17,and APCs via secretion of IL-35 and simultaneous induction of Treg amplification to promote the production of Bregs producing IL-35 and IL-10[173,174];
(3) Induction of anergic CD8+T cell and Treg development by secreting TGF-β [175];
(4)Inhibition of effector T cell expansion or induction of effector T cell apoptosis by expression of FasL and granzyme B [176,177];
(5) Overexpression of PD-L1 to inhibit follicular helper T cell(Tfh) differentiation and proliferation;
(6) Production of inhibitory IgG4 and sialylated IgG to limit inflammation [178].
Clinical studies have shown that patient’s with operational tolerance,such as kidney transplant recipients,have maintained graft function for many years without taking immunosuppressive drugs,and the absolute number and proportion of B cells are elevated in these patients compared with those in patients who experienced graft rejection, indicating that B cells may play a regulatory role in OTT [179,180]. Another study showed that, compared with patients taking immunosuppressive agents, the number of naive and transitional B cells was increased in peripheral blood samples.Meanwhile, B cells expressing CD20 detected in urine sediment were increased, and this increase in cell number was also noted in kidney transplant-tolerant patients who did not take immunosuppressants [181].
4. Clinical translation of cell therapies in transplantation
Adoptive cell therapy is a recently developed method used to promote allograft tolerance. The transfer of regulatory immune cells to recipients before or after transplant inhibits the activation of effector cells and promotes graft tolerance [182]. Most clinical trials carried out globally on the induction of graft tolerance by adoptive cell therapy in solid OT are still underway, and few reports have been published.
Adoptive Treg therapies can control acute and chronic rejection in many animal transplant models and may be used in humans.Results of TRACT, a phase I dose-escalation safety trial infusing ex vivo expanded recipient polyclonal Tregs into kidney transplant recipients, was published (NCT02145325) by Northwestern University (Chicago, USA) in 2018 [183]. Nine renal transplant recipients were divided into three groups, and infusion of 5 × 108, 1 × 109, and 5 × 109Tregs, respectively, was carried out at 60 days after transplantation. The Tregs were isolated from leukocytes,which had been collected one month before transplantation and expanded ex vivo for 21 days.During the follow-up period, no serious adverse events due to reinfusion of Tregs were observed to have occurred, and none of the recipients presented with opportunistic infections associated with non-specific immunosuppression. The number of Tregs in the Tregs-reinfused recipients was increased compared to that in the control group under the same immunosuppressive conditions. The presence of DSA was observed in two recipients due to drug intolerance or overt noncompliance.Overall,the trial demonstrated that it is safe to infuse ex vivo-expanded Tregs to kidney transplant recipients.The authors of the TRACT trial, therefore, are planning a phase II trial as well.
Hokkaido University Hospital and University of California(UCSF, USA) have completed and published clinical trials using the expanded recipient Tregs for reinfusion. Todo et al. [184] conducted a clinical trial wherein ten liver transplant recipients received ex vivo expanded ‘‘Tregs-enriched” cell product reinfusion,and seven subjects had successfully stopped immunosuppressants. Chandran et al. [185] conducted a clinical trial of UCSF for autologous polyclonal Tregs reinfusion in kidney transplant recipients. The results of the trial showed that the isolation, expansion,and reinfusion of Tregs are safe and feasible in transplant recipients taking immunosuppressive agents post transplantation(NCT02088931).
Currently,the European Union ONE Study is conducting a phase I/II clinical trial of autologous TolDCs for cell therapy in live kidney transplant recipients to assess their safety and feasibility(NCT02252055) [186]. Thomson et al. [187] have also proposed a phase I/II clinical safety trial investigating the effects of donorderived DCregs combined with conventional immunosuppressive agents on clinical and subclinical renal transplant rejection patients.Clinical trials of adoptive MSC therapy to induce immune tolerance in liver, lung, and kidney transplant patients have also been conducted[188–190].Perico et al.[191]showed that autologous MSCs could protect transplanted kidneys from graft dysfunction before renal transplant(NCT00752479).Although the number of clinical trials on adoptive Mreg treatment is limited, the results are promising. Two recipients required very low-dose tacrolimus monotherapy for stable renal function after six years of adoptive transfer therapy (NCT00223067) [192]. While all of the abovedescribed cell adoptive therapies show promise in terms of safety,feasibility, and tolerability, the route of administration, time of administration, the dose administered, and the optimal combinations of these therapies with other therapeutic modalities remain to be fully elucidated.
5. Application of chimeric antigen receptor technology in adoptive cell therapy
Currently, chimeric antigen receptor (CAR) T cell therapy has shown great potential in clinical anti-tumor applications, but its application in the induction of graft tolerance by immune cells(e.g.,Tregs)has so far been limited to the laboratory.Nevertheless,the technology holds promise in improving the antigen specificity of certain immune cells to induce tolerance. Recently, innovative CAR-Treg therapies in animal transplant models have been reported. CAR-Tregs genetically modified with coding CARs are non-MHC dependent and have better antigen specificity than the conventional Tregs. Hombach et al. [193] genetically modified Tregs using CAR technology a decade ago and engineered‘‘designer Tregs” with defined specificity. The mechanism of CAR-Tregs in inducing immune transplant tolerance is similar to that of polyclonal Tregs.Pierini et al.[194]constructed mAb-CAR-Tregs targeting specific tissue sites and successfully reduced GVHD in the mouse model. The mAb-CAR-Tregs targeted MHC class I proteins on allografts and prolonged the survival of islet allografts and secondary skin grafts.
Similarly, in the animal skin transplant model, Boardman et al.[195] and Noyan et al. [196] designed CAR-Tregs for HLA-A2 to induce immune tolerance. The designed CAR-Tregs could inhibit graft rejection more effectively than polyclonal Tregs. ANS8-CARTregs engineered for FVIII antibodies could also significantly inhibit the proliferation of FVIII-specific T-effector cells in hemophilia A patients. Thus, these Tregs have strong potential for further use in tolerogenic therapy of hemophilia A patients [197]. To date,there have been no reports on the implementation of CAR technology for induction of transplantation tolerance by other immune cells.
6. Conclusions
Although T cells are thought to be the main effector cells involved in OTR and OTT,the role of innate immune cells in transplant immune responses has received increasing attention in recent years.
Notwithstanding the different types of rejection in OT,rejection can be roughly divided into two categories: acute rejection and chronic rejection.Regardless of the type of rejection,precise preoperative tissue matching and appropriate desensitization treatments (e.g., plasma exchange, immunoadsorption, and high-dose intravenous immunoglobulin) are essential to prevent rejection.For acute rejection after transplant surgery, hormone pulse therapies, strong immunosuppressants, anti-human immunothymocyte globulin (ATG) or anti-human T lymphocyte immunoglobulin(ALG), plasma exchange, immunoadsorption, and intravenous immunoglobulin therapies should remain the default clinical treatment strategies.
Although there are no effective treatments for chronic transplant rejection, we posit that therapies utilizing adoptive immune cells to prevent and treat chronic transplant rejection will see widespread use and significantly improve patient prognosis in the years ahead. Efforts should be made to develop adoptive cell therapies with low immunogenicity and high universality and specificity, such as universal chimeric antigen receptor Tregs (UCAR-Tregs). Universal cell products should be used before surgery to improve the success rate of transplants through induction of immune tolerance.
Animal organs could be considered for use in human transplantation; however, immunogenicity caused by animal organs is a major hindrance to their use.Therefore,future studies should focus on directly examining these issues in the context of clinical needs,aiming to optimize cell product manufacturing methods (e.g., UCAR-Tregs), the associated cell product equipment (e.g., cell sorter), and specific cell product and cell quality control procedures for OT patients.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81971495 and 91442117), CAMS Innovation Fund for Medical Sciences (2019-I2M-5-035), the National Science Foundation of Jiangsu Province (BRA2017533 and BK20191490), the State Key Laboratory of Reproductive Medicine(SKLRM-K202001),and the Foundation of Jiangsu Collaborative Innovation Center of Biomedical Functional Materials.
Compliance with ethics guidelines
Xiaojie Gan, Jian Gu, Zheng Ju, and Ling Lu declare that they have no conflict of interest or financial conflicts to disclose.

- Engineering的其它文章
- Corrigendum to ‘‘Research on DC Protection Strategy in Multi-Terminal Hybrid HVDC System” [Engineering 7 (2021) 1064–1075]
- A Vision of Materials Genome Engineering in China
- Roles of Serum Amyloid A 1 Protein Isoforms in Rheumatoid Arthritis
- Ecological Barrier Deterioration Driven by Human Activities Poses Fatal Threats to Public Health due to Emerging Infectious Diseases
- Chimeric Antigen Receptors and Regulatory T Cells: The Potential for HLA-Specific Immunosuppression in Transplantation
- Review on Drug Regulatory Science Promoting COVID-19 Vaccine Development in China