
Chimeric Antigen Receptors and Regulatory T Cells: The Potential for HLA-Specific Immunosuppression in Transplantation
2022-06-11 09:04:20SabrinaWrightConorHennessyJoannaHesterFadiIssa
Engineering 2022年3期
Sabrina Wright, Conor Hennessy, Joanna Hester, Fadi Issa*
Nuffield Department of Surgical Sciences, University of Oxford, Oxford OX3 9DU, UK
Keywords:Chimeric antigen receptors T cell Treg Alloimmunity Bioengineering Transplant Autoimmunity
ABSTRACT Chimeric antigen receptors(CARs)are a breakthrough in genetic engineering that have revolutionized the field of adoptive cellular therapy(ACT).Cells expressing these receptors are rerouted to a predefined target by the inclusion of an antigen-specific binding region within the synthetic CAR construct.The advantage of cells with programmed specificity has been demonstrated clinically in the field of oncology,and it is clear that such cells have greater accuracy, potency, and reduced off-target therapeutic effects compared with their unmodified counterparts.In contrast to conventional T cells(Tconvs),regulatory T cells(Tregs) play a major role in suppressing immune activation and regulating the host immune response.CAR expression within Tregs has been proposed as a therapy for autoimmune and inflammatory diseases,graft-versus-host disease(GVHD),and organ transplant rejection.In the latter,they hold immense potential as mediators of immune tolerance for recipients of allotransplants. However, current research into CAR-Treg engineering is extremely limited, and there is uncertainty regarding optimal design for therapeutic use. This review examines the rationale behind the development of CAR-Tregs, their significance for human transplantation, potential designs, safety considerations, and comparisons of CAR-Tregs in transplantation models to date.
1. Introduction
Chimeric antigen receptors (CARs) are a breakthrough in the design and production of cells for adoptive cellular therapy(ACT).CAR-T cells expressing such receptors are rerouted to a predefined target by the inclusion of an antigen-specific binding region within a synthetic construct. The advantage of cells with programmed specificity has been demonstrated in numerous xenograft models, and it is clear that antigen-specific cells have greater accuracy, potency, and reduced off-target therapeutic effects compared with their unmodified counterparts. The induction of CAR expression within regulatory T cells (Tregs) has been put forward as a putative therapy for autoimmune and inflammatory diseases, graft-versus-host disease (GVHD), and organ transplant rejection (Table 1) [1–5]. CAR-Tregs hold immense potential as mediators of tolerance in the recipients of allogeneic transplants and therefore deserve greater attention. A review at this stage is necessary to both highlight the need for, and encourage greater research effort in,this field and to identify areas of missing information that must be addressed before experimental research can progress to clinical applications.
For the purpose of this review,Tregs are defined as CD4+CD25+-FOXP3+(CD: cluster of differentiation; FOXP3: forkhead box P3) T cells; conversely, the CD4+CD25–FOXP3–subset of T cells with proinflammatory properties are referred to herein as conventional T cells (Tconvs). When describing T cells that have been modified to express a CAR, modified Tregs are referred to as CAR-Tregs,whereas modified Tconvs are referred to as CAR-Tconvs.
1.1. Mechanisms of Treg suppression
In 1995,Tregs were formally recognized as a distinct subset of T cells by Sakaguchi et al.[6],who identified them by the marker signature CD4+CD25+. This landmark paper illustrated the ability of Tregs to suppress allogeneic responses and revealed that depletion of Tregs led to a heightened immune response and the spontaneous development of autoimmune disease. Later work identified the transcription factor FOXP3 as a defining regulator of the Treg phenotype [7]. Tregs are a naturally occurring subset of lymphocytes in the adaptive immune system that are responsible formaintaining immune homeostasis. In addition to mediating tolerance for self-antigens, they act under inflammatory conditions to limit the effector immune response in order to prevent excessive damage to an individual’s tissues. Tregs manipulate the immune environment through a variety of contact-dependent and soluble mechanisms. They are able to directly suppress other subsets of immune cells, including B cells and CD4+CD25–Tconvs, and act on dendritic cells(DCs)to prevent maturation and antigen presentation [8,9]. The following are major strategies employed by Tregs to moderate the immune system in response to antigen stimulation.
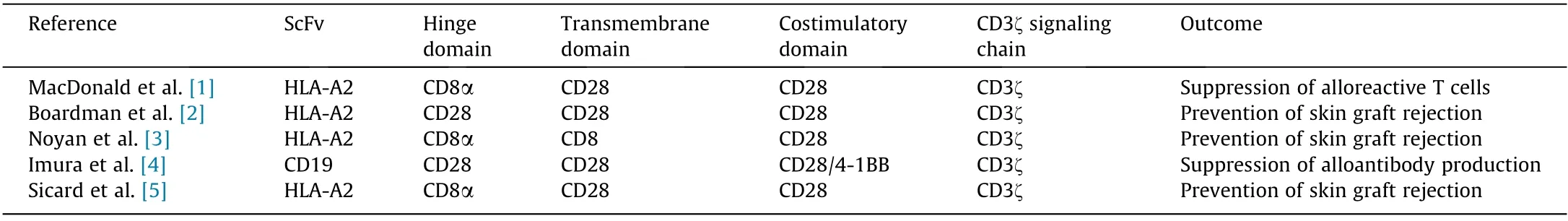
Table 1 Summary of five current studies examining whether CAR-Tregs can modulate the alloimmune response.
1.1.1. Cytolysis
Cytolytic molecules such as granzyme B are released to instigate the direct killing of target proinflammatory cells. Granzyme B production by thymus-derived natural Tregs (nTregs) induces apoptosis in Tconvs and B cells [10,11]. The perforin/granzyme pathway mediates direct cytotoxicity, but the relationship between perforin and granzyme B has been somewhat controversial. Two in vitro studies have described a granzyme Bdependent, perforin-dependent suppression system by FOXP3+cells expressing granzyme B [12,13], whereas Gondek et al. [10]reported independence between the two systems.Using granzyme B-deficient mouse models, Cao et al. [14] reported that perforin and granzyme B are both important for the Treg-mediated apoptosis of natural killer(NK)and CD8+T cells,since the adoptive transfer of Tregs lacking the gene for either of these molecules failed to suppress tumor clearance.Similarly,perforin was found to be necessary for Treg-mediated removal of DCs in a perforin knockout model [15].
1.1.2. Cytokine release
Interleukin (IL)-10 and IL-35 are key regulatory cytokines responsible for suppressing proinflammatory responses [16]. IL-10 prevents the release of proinflammatory cytokines and chemokines by Tconvs,while inhibiting the expression of costimulatory molecules and class II major histocompatibility complexes(MHCs) on DCs and other professional antigen-presenting cells(APCs) [17–19]. IL-35 is a relatively recent addition to the known repertoire of Treg tools and has since been shown to directly inhibit the proliferation of Tconvs in mice [20]. Although IL-35 is not constitutively expressed in human Tregs, long-term activation of Tregs in humans leads to the upregulation of IL-35,thereby conferring an enhanced suppressive capacity [21,22].
Interestingly, ex vivo-induced Tregs (iTregs) appear to rely on cytokine signaling rather than cytotoxicity for their function;several studies have reported that transforming growth factor β(TGF-β) signaling by iTregs is responsible for the suppression of B cells, T cells, and DCs [11,23]. However, thymic Tregs have been favored over iTregs in preclinical models to date(likely due to their greater stability in vivo), so an in-depth analysis of iTreg mechanisms is outside the scope of this review.
1.1.3. Manipulation of DCs
Tregs are able to weaken or abrogate activation signals from professional APCs to prevent the activation of naïve T cells [24].The Treg molecule cytotoxic T lymphocyte antigen 4 (CTLA-4)engages with the costimulatory molecules CD80 and CD86 on the APC cell surface,triggering the release of indoleamine 2,3-dioxygenase(IDO),a tolerogenic enzyme that acts as a rate limiter for tryptophan catabolism in Tconvs and results in an increased concentration of tryptophan metabolites in the surrounding milieu,which have suppressive effects on Tconv cell cycle progression [24,25]. In addition, T cell receptor (TCR) binding stimulates the downregulation of CD80 and CD86 on DCs in a CTLA-4-dependent manner [24].
1.1.4. Metabolic disruption and competition
Metabolic disruption through competition for essential cytokines is a further potent mechanism of suppression. In particular,Tregs can deprive surrounding cells of IL-2 directly by consuming the external supply, thereby limiting the growth and survival of non-Tregs [26]. When aggregated in a single region, the high expression of CD25 (IL-2 receptor α) on Tregs may bind enough IL-2 to induce apoptosis among the surrounding cells [26].
1.1.5. Infectious tolerance
Finally,Tregs may induce a tolerogenic phenotype in inflammatory cells through a phenomenon known as infectious tolerance[27,28]. This requires membrane-bound TGF-β on the surface of Tregs,which induces the de novo generation of FOXP3+T cells from naïve precursors in a contact-dependent manner [29]. IL-35 has the ability to convert target Tconvs into Tregs;thus,it may be considered an agent of infectious tolerance [22].
1.2. Tregs as an emerging therapy
The therapeutic strategy of adoptive cellular therapy(ACT)harnesses the intrinsic function of immune cells; in the case of Tregs,this is the promotion of a state of tolerance.Further characteristics of Tregs, including their ability to proliferate, interact with other cell types, and exert multiple suppressive mechanisms, support the superiority of Treg therapy over non-cellular approaches.
It was realized soon after the discovery of Tregs that their suppressor function could be exploited to inhibit the immune response in autoimmune diseases and in the context of transplantation [30,31]. This notion was followed by in vivo studies on the effectiveness of Treg transfer therapy against various autoimmune diseases (including type 1 diabetes, experimental autoimmune encephalomyelitis, and systemic lupus erythematosus), GVHD,and allogeneic transplantation [32–36]. It was shown that ex vivo expanded Tregs could prolong skin and islet allograft survival in humanized mouse models by reducing CD4+and CD8+T cell infiltration,generating optimism that these results could be replicated in human patients [37,38].
The concept of using Tregs in transplantation has gained traction principally due to the toxic side effects of non-cellular immunosuppressive drugs, and it is hoped that cell-based therapy will mitigate the risk of toxicity[39,40].Higher incidents of cardiovascular disease and diabetes are observed in transplant recipients; although these conditions are greatly influenced by the comorbidities of the patient prior to transplantation, they are also directly affected by pharmacologic immunosuppressive agents[39,41]. Hypertension is commonly reported in renal transplant patients,chiefly as a result of treatment with calcineurin inhibitors,and its prevalence can reach as high as 82% in adult patients[39,42]. These factors in turn elevate the risk of cardiovascular morbidities such as myocardial infarction, cardiomyopathy, heart failure, and stroke [40]. Overall, organ toxicity severely impacts patient quality of life and remains a major cause of both graft loss and patient death in transplant recipients [40].
Cell therapy may also be of greater practicality for patients who would otherwise follow a daily regimen of medication.Unmodified polyclonal Tregs may remain detectable in humans from two weeks up to one year,allowing for a far greater timespan between doses; this would be less burdensome for the patient and reduce the chance of allograft rejection resulting from non-adherence[1,43,44]. A question that remains to be answered is whether repetitive dosing of Tregs is necessary, or whether it is possible to establish a self-sustaining population of donor-specific Tregs from limited infusions.
The first in-human clinical trials of Treg adoptive transfer gave evidence to support the use of expanded polyclonal Tregs for the prevention or treatment of GVHD and autoimmune diseases[45–47].In accordance with similar studies in mice[48],early Treg infusion in human patients was shown to prevent chronic GVHD in the absence of any concurrent immunosuppression;moreover,Treg therapy was associated with greater cellular immunity against opportunistic pathogens while preserving the graft-versusleukemia effect[49].However,the mortalities highlighted the need for improvement in terms of safety: Patients remained vulnerable to adenoviral infection,toxoplasmosis,bacterial infection,and fungal infection from a generically suppressed immune system.
More recently, solid organ transplantation became the subject of a phase I dose escalation trial to assess the safety of infused Tregs in kidney transplant patients. In this trial by Mathew et al.[50], nine recipients of living-donor kidney allografts received ex vivo expanded autologous Tregs at two months posttransplantation, following lymphocyte depletion and in combination with mycophenolate mofetil (MMF) and sirolimus maintenance therapy. It was of major interest that no serious adverse events were reported, and no evidence of opportunistic infections or other immunosuppression-related conditions was seen. The researchers reported 100% graft survival at two years post-transplantation [50]. Further research is ongoing to establish the potential of Treg treatment with minimal concurrent immunosuppression.The ONE Study is an international consortium that was assembled to evaluate different regulatory cell types in the context of living-donor renal transplantation, with Tregs featuring among these [51]. Patients were initially given triple therapy (prednisolone, MMF, and tacrolimus) and later infused with autologous polyclonal Tregs at five days post-transplantation [52]. Out of 38 patients receiving regulatory cells who completed the observation period,15 were successfully weaned onto tacrolimus monotherapy and exhibited fewer viral infections compared with patients on standard immunosuppressive therapy [51]. A follow-up phase II trial entitled the TWO Study was later announced, which aimed to assess the efficacy of Tregs in combination with sirolimus monotherapy in preventing renal transplant rejection [53].Although it is unclear at this stage whether Tregs could entirely replace conventional drugs,even the reduction of immunosuppression down to a monotherapy would have significant positive consequences for the outcome of the allograft and the patient.
1.3. Antigen-specific Tregs
Non-specific suppressors of the immune activation pathway,such as metabolic inhibitors,confer systemic suppression such that the entire body—and not merely the donor organ—is affected. In addition to cardiotoxicity and nephrotoxicity, heavy regimens of systemic immunosuppression are known to increase a patient’s risk of cancer—especially of squamous cell carcinoma and Kaposi sarcoma [54]. Reducing the patient’s ability to detect and respond to pathogens also renders them especially vulnerable to bacterial,fungal,viral,and mold infections[55].While cellular therapies circumvent many of the toxicities associated with pharmacologic drugs, the risk remains that Tregs bearing polyclonal specificity will still increase vulnerability to the development of infection and malignancies in transplant patients. To combat this issue,novel therapies are being designed to target antigens specific to the donor tissue, so that other organs expressing self-antigen are not affected.
While designers of cancer-specific CAR-Tconvs face the difficulty of selecting a suitable target (there are few truly cancer-specific antigens),in the case of transplantation,there will be discrepancies between the expressed allelic repertoire of the human leukocyte antigens(HLAs)of the donor and the recipient.This makes it possible to target antigens that are unique to the donor organ and absent from the recipient,enabling high specificity for the allograft.
Tregs represent less than 10% of the circulating CD4+T cell population, and only a small proportion of these will bear the correct TCR for an antigen of interest [56–58]. It is common to selectively expand antigen-specific Tregs in vitro by co-culturing bulk Tregs with allogeneic DCs, or with autologous DCs that have previously been pulsed with allopeptide [57,58]. Alternatively,genetic manipulation can be used to directly confer specificity to a polyclonal cell sample. Many preclinical studies have cloned recombinant TCRs of known specificity into host CD4+T cells for the purpose of creating a targeted cell therapy;this has been done in Tregs as well as in Tconvs [59–61], and promising results have been obtained from experimental models of multiple sclerosis and type 1 diabetes [60,61]. However, although MHC processing by TCRs allows for the recognition of intracellular (IC) peptides,there is also an argument that a major disadvantage of TCRs,whether endogenous or modified,is that they are restricted to peptides associated with an MHC,limiting the pool of target antigens.Moreover, evidence for the downregulating effects of calcineurin inhibitors on MHC expression in DCs suggests that the process may be compromised if the patient is receiving certain pharmacologic immunosuppression [62,63].
In answer to these problems, the development of the first CAR was reported in 1989 by Gross et al. [64]. This receptor combined the constant and variable domains of an anti-2,4,6-trinitrophenyl antibody with a segment of an α or β TCR chain;the total construct was then transfected into a cytotoxic T cell hybridoma.The resulting transgenic cells were demonstrated to have antigen-specific,non-MHC-restricted effector functions, as well as the ability to induce cytokine production in their target cells. By using the variable fragment of an antibody as opposed to the MHC-restricted binding region of a TCR, the synthetic receptor could engage with extracellular (EC) peptides without the need for processing.Importantly for allogeneic transplantation, this meant that MHC molecules themselves could also be targeted.
1.4. Development of CAR designs
Following on from the revolutionary ‘‘first-generation” CAR of Gross et al.[64],many other researchers have expanded the design and application of CARs to generate improvements in longevity and functionality. CARs have been engineered into various cell types, including NK cells, CD8+T cells, Tconvs, and Tregs [65–69].
CARs are an improvement on the TCR design in that they are modular, and individual segments of the CAR construct may be added or substituted(Fig.1).For example,a costimulatory domain may be formed from different domains of the TCR—with CD28 and 4-1BB (CD137) being the most commonly used—and each confers its own advantages or disadvantages on the functionality of the CAR. Additional components, such as a cytokine sequence, may also be incorporated to enhance their function.
CAR therapy has shown remarkable efficacy in the treatment of relapsed or refractory cancer where previous lines of treatment have failed.Their profound success in preclinical models,and later in clinical trials, paved the way for the approval of two independent CAR-Tconv therapies by the US Food and Drug Administration(FDA)in 2017,followed by approval from the European Medicines Agency (EMA) a year later [70]. One of these therapies, tisagenlecleucel (marketed as Kymriah®), achieved a 93% complete remission rate in an early phase I/IIa trial involving 75 patients [71].CAR-Tregs have similarly demonstrated efficacy in treating autoimmune and inflammatory diseases in experimental murine models, as well as in preventing GVHD in models of allogeneic stem cell transplantation,and have demonstrated superior efficacy in allogeneic models of solid organ transplantation compared with polyclonal Tregs [1,2,72–76] (Fig. 2).
The production of CAR-Tregs has been a very recent milestone in the development of genetically engineered T cells. The first in vivo CAR-Treg study to investigate their relevance in allogeneic transplantation was published in 2016 by MacDonald et al. [1],who illustrated the ability of CAR-Tregs to confer antigen-specific suppression against an HLA; yet their potential as a prophylactic and treatment for transplant rejection remains highly underresearched. No human trials of CAR-Tregs have yet taken place and, consequently, there are no approved CAR-Treg therapies.Nevertheless, the recent approval of the anti-CD19 CAR-Tconvs Kymriah®and Yescarta®by the FDA and the EMA offers encouragement that CAR-Tregs may similarly gain consent for clinical applications in the near future [70].
2. Designing an effective CAR for therapeutic Tregs
The first designs to employ basic chimeric molecules with an antigen-specific binding region—which have since been labeled‘‘first-generation” CARs—showed very limited persistence in vivo[77]. Subsequent designs have improved upon the expansion and persistence of the archetype (Fig. 3). Second-generation CARs include a costimulatory domain between the transmembrane domain and the CD3ζ signaling domain to allow for full activation of the cell; this markedly improves the expansion and, consequently,the efficacy of both Tconvs and Tregs bearing the receptor[77–80]. Third- and fourth-generation CARs have also been developed to incorporate additional elements. All CARs, regardless of generation,contain a CD3ζ chain at the IC tail to propagate the activation signal, mimicking activation through the TCR. While excellent reviews have already been published on the design of CARs,the importance of each component in relation to CAR-Tregs specifically will be discussed here.
2.1. Selecting an HLA target
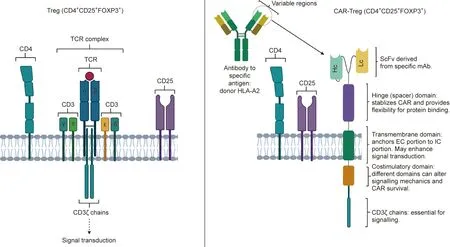
Fig. 1. Conventional Treg versus CAR-Treg design. CAR-Tregs retain the key components of Tregs: the CD25 and CD4 complexes. The TCR complex is replaced with the modular CAR-Treg design,which provides antigen specificity depending on the monoclonal antibody(mAb)from which the single-chain variable fragment(ScFv)is derived.This allows the production of Tregs with targeted anti-inflammatory effects,which may have applications in downregulating the alloresponse and preventing graft rejection in allogenic transplants. Hc: heavy chain; Lc: light chain.
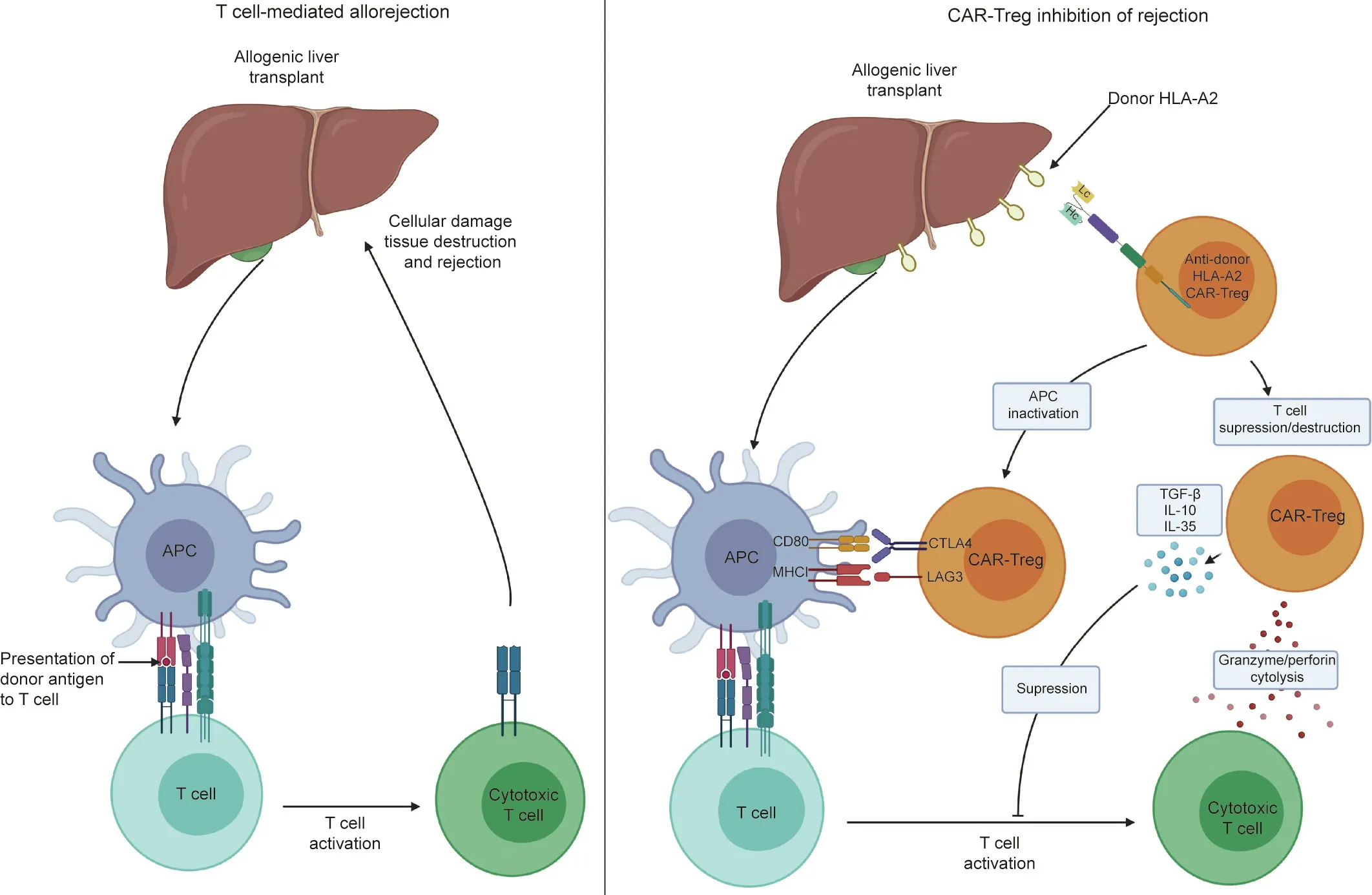
Fig.2. Proposed mechanism of CAR-Treg-mediated suppression of alloimmunity.Organ rejection following allotransplantation is traditionally thought to be mediated by T cell-mediated alloimmunity.APCs such as DCs present antigen from the donor tissue to host T cells,which are then activated by the recognition of‘‘non-self.”This ultimately leads to the production of cytotoxic T cells,which propagate tissue destruction and hence organ rejection.The proposed function of CAR-Tregs would potentially prevent this.The construction of a CAR that recognized an antigen on the donor organ(e.g.,HLA-A2)would result in Treg activation and proliferation.These CAR-Tregs could then directly inactivate APCs, acting via cytotoxic T-lymphocyte-associated protein 4 (CLTA4)–CD80 and major histocompatibility complex class I (MHCI)–lymphocyte activation gene 3 products (LAG3) interactions, thus preventing the presentation of donor antigen to T cells. Furthermore, the CAR-Tregs could directly inhibit T cell activation via the production of inhibitory cytokines(TGF-β,IL-10,and IL-35).Finally,the activated CAR-Tregs could inhibit rejection by destroying cytotoxic T cells via the release of granzyme and perforin.
Within the CAR, antigen specificity is typically determined by the inclusion of the light and variable chains of a single-chain variable fragment (scFv) derived from a monoclonal antibody (mAb).To date, all CAR-Treg allograft models have used the MHC class I HLA-A2 antigen as the therapeutic target, for the reason that it is commonly found across human populations and therefore a large number of patients can be treated with a single CAR design[1–3,5,81–90].This is preferable in order to reduce the labor,costs,and time required to produce and expand each clone; from a clinical perspective, CAR-Tregs could be taken ‘‘off the shelf” as required for delivery into patients. However, other common HLA antigens should be explored as potential targets for CAR-Treg therapy in order to cover the various haplotypes present throughout populations. Indeed, if CAR-Tregs are truly envisaged as a future therapeutic agent, then the variation of haplotype frequencies across geographical and ethnic groups requires an expanded panel of antigen targets [83–90].
Preclinical models of CAR-Tregs have not specified to which subtypes of HLA-A2 the scFv can bind, out of over 800 that are currently known [81,82,91]. It would be convenient to produce CARs that can target multiple alleles to reduce the number of CAR designs required to cover all patient HLA haplotypes,although this scenario would need careful controlling to ascertain that no unwanted cross-reactivity takes place between donor and recipient (a safety concern that will be discussed further on).Evidence suggests that HLA-binding peptides can react with multiple different subtypes [92].
2.2. Costimulatory domains
CD28 and 4-1BB are most commonly used as the costimulatory element of the CAR construct[1–3,5,80,93].They each demonstrate different effects on the signaling kinetics and persistence of the CAR, and even on the suppressive function of the host cell [94].The IC signaling strength of CD28 is higher than that of 4-1BB;consequently,there is a greater cellular expansion rate,which renders the host cells more susceptible to exhaustion and is associated with poorer persistence[95,96].Conversely,4-1BB CARs have been demonstrated to survive in humans for over 600 days after a single infusion [71].
The first study to directly compare the effects of CD28 versus 4-1BB on the phenotype and function of CAR-Tregs reported that CD28 is superior to 4-1BB at promoting cytokine secretion and overall suppressive capacity [94]. CAR-Tregs bearing the CD28 domain produced significantly more IL-10 than their 4-1BB counterparts;this was associated with a decreased suppressive capacity among 4-1BB CAR-Tregs when co-cultured with CD4+and CD8+Tconvs in vitro. Furthermore, the study reported that 4-1BB even decreased the suppressive function of CAR-Tregs, compared with first-generation CARs lacking any costimulatory domain.
Other costimulatory domains that have been less extensively tested in CARs include inducible T-cell costimulator (ICOS,CD278), tumor necrosis factor receptor superfamily member 4(OX40),CD27,and CD40 ligand(CD40L)[97,98].It would be useful to gain more data on the efficacy of these domains and their functional effects on the Treg subset specifically.CD27 is a particularly hopeful candidate for enhancing persistence; Song et al. [99]demonstrated that CARs bearing a CD27 domain possess increased longevity in vivo compared with CARs bearing CD28.
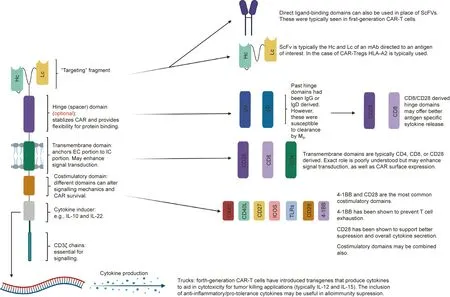
Fig. 3. The modular structure of CARs. CAR structure can be modified depending on the target and function of interest. The ScFv offers antigen specificity and is usually derived from mAbs directed at the antigen of interest. Hinge domains are an optional component that can increase cytokine production, enhance proliferation, or facilitate transmigration. Transmembrane domains anchor the EC component to the IC component and may also play a role in signal transduction. Costimulatory domains can affect signal transduction, increase cellular expansion rate, and prevent T cell exhaustion. They can be used alone or in combinations, depending on the desired effect. Fourthgeneration CAR-T cells have introduced transgenes that produce cytokines that can aid in the suppressive or cytotoxic effector actions of the cell,depending on its purpose.IgG: immunoglobulin G; IgD: immunoglobulin D; OX40: tumor necrosis factor receptor superfamily member 4; ICOS: CD278; CD40L: CD40 ligand; Mφ: macrophages;TLRs: Toll-like receptors.
Improvements upon the second-generation design have been attempted in third-generation CARs by including an additional costimulatory domain. A study by Ramos et al. [100] (using Tconvs)found that the combination of CD28 and 4-1BB overcame the limitations of each individual domain, resulting in greater persistence and expansion compared with second-generation CARs with CD28 alone.Once again,this subject has been untested in the field of CAR-Tregs and deserves investigation for its potential.
2.3. Cytokine domains
Further enhancements of the CAR construct have resulted in fourth-generation CARs incorporating a cytokine domain—most commonly IL-12 or IL-15—to enhance the cytotoxic effects of CARs engineered for anti-tumor immunotherapy [101–103]. Taking inspiration from this design and applying it to CAR-Tregs, the incorporation of regulatory cytokines such as IL-10 or TGF-β within CARs could potentially modulate the immune environment within the allograft. It would be clinically relevant to discover to what extent their inclusion improves the success of CAR-Treg therapy,in terms of creating a tolerogenic environment and preventing graft damage with minimal support from pharmacologic agents.
3. Gene-delivery systems
Different genome-editing techniques are associated with different efficiencies, specificities, expression stabilities, and safety profiles of CAR-expressing cells,which ultimately affect the success of the final CAR-Treg product.In this section,established methods for the genetic delivery of CARs are evaluated for their suitability in the context of CAR-Treg therapy.
3.1. Viral vectors
Viral transduction is the most popular approach for genetic manipulation of T cells.This can be attributed to their high editing efficiency and the ultimate incorporation of their DNA cargo into the host cell genome, resulting in stable protein expression [104].
Average CAR expression on retrovirally transduced Tregs ranges from 20%to 95%[3,76].While several CAR products manufactured via retroviral delivery have proceeded into clinical trials (notably,the FDA-approved Yescarta®)[105,106],there is evidence that such cells can express immunogenic vector-encoded epitopes, presenting a safety concern that viral vectors may increase the immunogenicity of therapeutic cells [107]. The risk of genotoxicity is enhanced through the semi-random integration pattern with bias toward transcription start sites [108,109]. Lentiviruses, a complex subfamily of the retrovirus, have gained popularity as a viral vector, and CARs engineered using this method have also progressed to clinical trials and beyond, including the FDA-approved Kymriah®[110–112]. CAR-Treg studies have reported differing ranges of lentiviral transduction efficiency, from 10%–15%(Fransson et al. [72]) to 30%–80% (Boardman et al. [2]). Lentiviral systems appear to be less genotoxic due to their tendency to favor active gene sites over transcription start sites, and are considered to pose a lower risk of insertional mutagenesis; therefore, they are more favorable in clinical applications [108,109,113].
Although CAR-Tregs have not yet entered clinical trials, CARTconvs may provide insight into their persistence in humans after viral transduction.Data from the ZUMA-1 clinical trial of axicabtagene ciloleucel (Yescarta®) demonstrated that CARs may remain readily detectable in peripheral blood for 180 days, and even up to 24 months in some patients; in this case, a CD28 domain was used [114]. Even more impressively, tisagenlecleucel (Kymriah®),a CAR therapy incorporating the 4-1BB domain,has been reported to persist in the blood for a maximum of 617 days, with a median persistence value of 168 days [71]. This evidence would suggest that viral delivery has the potential to support CAR expression for extensive and clinically appropriate time periods.
Paradoxically, CAR-Treg data from mouse models have not yet demonstrated such a lengthy duration of CAR expression using viral vectors.Data from mouse studies revealed CAR-Tregs surviving just two weeks after lentiviral transduction (MacDonald et al.[1]),over 40 days for retroviral transduction in an allotransplantation model(Noyan et al.[3]),and 17 weeks for retroviral transduction in a type 1 diabetes model by Tenspolde et al.[75].MacDonald et al.[1]hypothesized that persistence could be hindered by insufficient IL-2,insufficient antigen,or a combination of both;the first may certainly be a potential factor, as Tregs do not produce endogenous IL-2,instead relying on it being secreted by other cells.On the other hand,a comment by Noyan et al.[3]raised the point that mice were reconstituted with HLA-A2 peripheral blood mononuclear cells(PBMCs)in MacDonald’s study[1],meaning that broad activation of HLA-A2 CAR-Tregs would have taken place; in this case, the Tregs would experience greater exhaustion, leading to their rapid decline.Across these studies,the question of whether CAR-Tregs became exhausted due to the continuous presence of target antigen,or whether immunogenicity conferred by viral vectors was responsible for the decline of CAR-Treg populations,remains to be answered; these factors and their implications on the success of CAR-Treg therapy will be discussed later in this review.
The aforementioned viral vectors can transport DNA cargo of up to 8 kilobases(kb),well above the size of the average CAR sequence[115].However,gene integration occurs in a semi-random manner;this can lead to heterogeneous CAR expression and,potentially,the integration of the DNA sequence into a proto-oncogenic site,prompting some researchers to explore alternatives.
3.2. Transposons
Transposons are a non-viral means of genetic modification offering long-term expression and higher DNA cargo limits,although the risk of integration near proto-oncogenic sites is similar to that of viral vectors [116]. A transposon element is typically delivered into the cell, along with a transposase, via two separate expression plasmids (the transposase may also be delivered as messenger RNA (mRNA)) [117,118]. The transposase excises the cargo DNA and ‘‘pastes” it into the host genome in a semi-random manner [116].
The most important advantage of this method is the cargo capacity,which is a limiting factor in the efficiency of other methods; for example, the expression system PiggyBac can carry a cassette of up to 14.3 kb without compromising efficiency[117,119]. The expression efficiency of transposon systems is around 50% for human peripheral blood cells [120]. Transposonmediated CAR expression has previously been utilized in the clinical setting: as part of a phase I trial conducted by Kebriaei et al.[121], anti-CD19 CAR-Tconvs were generated ex vivo from patient-derived T cells using the hyperactive ‘‘sleeping beauty”(SB)system SB11 and were subsequently infused into 19 recipients of allogeneic stem cell transplants.The CAR transgene was detectable in peripheral blood for an average of 51 days and a maximum of 180 days. This example is an encouraging starting point in the pursuit of long-term CAR expression, and overall,the cargo capacity, expression efficiency, and persistence in vivo associated with transposon-based delivery are all advantageous for the manufacture of CAR-Tregs.
3.3. CRISPR-Cas9
The clustered regularly interspaced short palindromic repeats(CRISPR)-CRISPR-associated protein 9 (Cas9) system, abbreviated to CRISPR-Cas9, offers a highly precise method of genome editing.Because the target site for gene insertion can be defined by the sequence of the guide RNA, aberrant gene insertion is greatly reduced compared with non-specific vectors. CRISPR editing has a relatively low efficiency that can be problematic for large sequences such as CARs, although its extremely high specificity renders it an attractive method nonetheless.
Targeting the CAR sequence to a defined region of the genome enables modification of a specific cell type; it is therefore logical during the production of CAR-Tregs to target delivery to a gene that is common or unique to Tregs.Targeting the T cell receptor α constant (TRAC) gene reportedly generates uniform CAR expression across transfected cells, reduces tonic signaling, and results in the internalization and re-expression of the CAR following antigen exposure to delay exhaustion and prolong cell survival[122];however, the issue arises that contamination with proinflammatory T cell subsets is likely to occur in the resulting CAR+cell product unless the CD4+CD25+FOXP3+population is successfully isolated from the bulk T cell population prior to gene editing. Other knock-in targets that are more exclusive to the Treg subset should be explored, such as FOXP3.
There is concern that homology-directed repair (HDR)-mediated gene insertion is disruptive to the endogenous target gene.This problem has been addressed,however,by studies utilizing intron-targeting and homology-independent integration to preserve the endogenous sequence [123,124]. By using CRISPRCas9 technology to create double-stranded breaks in the donor plasmid and the target gene, which share the same single guide RNA (sgRNA) target, the donor template can be efficiently integrated in a non-homologous manner without disrupting expression of the endogenous target gene [124]. This offers a superior approach for CAR insertion and makes the targeting of truly Treg-specific genes, such as FOXP3, a feasible option [122,123].
4. Source of cells
4.1. Autologous
Autologous Tregs express the MHC repertoire of the patient and are therefore not inherently immunogenic, making them a safer and more viable option for therapeutic use. Cells must be isolated several weeks ahead of infusion to allow time for genetic modification and expansion, and the generation of autologous CAR-T cells takes between 14 and 51 days [105]. It must be remembered that the objective of Treg therapy for transplantation is to circumvent the accumulative risk associated with long-term pharmacologic immunosuppression,and that short-term use of the standard triple therapy immediately following transplantation during the period of Treg expansion may not compromise the purpose of Treg therapy.Clinical trials of non-transgenic Tregs have performed delayed infusion after surgery, until which time the patient has been receiving medication that is gradually tapered after receiving the autologous cells [52,125].
4.2. Allogeneic
The use of third-party Tregs for CAR therapy possesses clear advantages and disadvantages.On the one hand,it has been envisaged that using cells from a third-party donor will allow for cell cultures to be expanded and banked,to be made available for multiple patients in a convenient ‘‘off-the-shelf” scenario when required. Furthermore, the cells can be extensively screened for quality prior to use [126]. Mouse models have indicated that the adoptive transfer of allogeneic Tregs into fully MHC-mismatched recipients can prevent the rejection of allografts bearing the same MHC profile as the Treg donor [127]. On the other hand, the immunogenic potential of allogeneic cells can have a detrimental effect on their long-term persistence.Kebriaei et al.[121]reported a significantly shorter detection period for infused allogenic CAR-T cells compared with autologous CAR-T cells bearing the same specificity (a maximum of 180 versus 360 days, respectively) in human hematopoietic stem cell transplant (HSCT) recipients; this result highlights the complications arising from the immunogenic nature of such cells (the researchers attributed the decreased survival to their use of concomitant immunosuppression to control GVHD and to the lack of lymphodepletion in their protocol). To reduce immunogenicity, further cell modification could be employed to abolish the expression of MHC and thus avoid immune clearance[128].While this potential solution could allow for many transplant patients to be treated promptly with CARTregs of a single donor-derived origin, many different specificities would have to be generated to cover the vast array of MHC haplotypes across the population. With this in mind, it is likely that autologous cells will be the preferred source of Tregs for the immediate future of ACT.
5. Adverse effects and safety issues
Despite the clear therapeutic benefits of CAR therapy and its clinical approval for certain malignancies, the technology is still in its fledgling stage. There are many unanswered questions,theoretical risks, and side effects in the literature that must be addressed. Numerous safety issues present an obstacle to the application of CAR-Treg therapy to human transplant patients, especially where a vital organ is the target of immunosuppression. At this stage, the published literature on the behavior, efficacy, and safety of CAR-Tregs in vivo remains extremely limited.
The following section lists important safety concerns highlighted in the current relevant literature and offers possible solutions.
5.1. On-target cytotoxicity
Granzyme B production has long been known as a mechanism of Treg-mediated suppression. This is problematic in the context of transplantation,as the donor organ becomes vulnerable to cytotoxic damage upon the delivery of tolerogenic cells [10,94]. The findings of Boroughs et al.[94]suggest that CAR-Tregs are capable of inducing apoptosis in cells expressing target antigen via the granzyme B/perforin pathway. Although cytotoxicity—measured by epithelial destruction and apoptosis—was minimal in their study, their results warrant investigation into how destructive mechanisms of CAR-Treg suppression may be mitigated. The researchers reported inhibition of granzyme B production when CAR-Tregs were treated with the phosphatidylinositol 3-kinasemammalian target of rapamycin (PI3K-mTOR) pathway inhibitor,rapamycin (sirolimus), during culture, which reflects previous findings on the effects of PI3K-mTOR pathway inhibitors on granzyme B [129]. The researchers alternatively suggested knocking out the GZMB gene;the effects of this genetic modification strategy would then persist in vivo without the need for the repeated administration of an mTOR inhibitor [94].
Another straightforward approach for reducing cytotoxicity is to reduce the number of cells per infusion; however, this necessitates a proficient understanding of the minimum cell number required to mediate tolerance toward the allograft.At the moment,it is unknown what the optimum dosage is for antigen-specific CAR-Tregs in solid organ transplantation.
On-target, off-organ binding is another concern after the adoptive transfer of cells. The expression of the target antigen on therapeutically irrelevant cells presents a substantial obstacle in the field of CAR-T cell therapy for malignancies [130]. Yet, unlike cancer immunotherapy, which often relies on the overexpression of tumor-associated antigens that are present to a lesser extent on healthy tissue,CARs in a transplantation setting can be targeted to a donor HLA allele that is not expressed by the recipient,reducing the risk of toxicity resulting from the erroneous binding of CARs.
5.2. Cross-reactivity with other peptides
Off-target toxicity is a theoretical risk with CAR-Tregs, principally due to the tendency for HLA-specific peptides to cross-react with other HLA subtypes [92]. Noyan et al. [3] performed a crossreactivity test for their HLA-A2-specific CAR-Tregs by exposing them to PBMCs of 20 different HLA haplotypes; while the CARs preferentially bound to HLA-A2, there was one recorded instance of cross-reactivity with HLA-A1-typed PBMCs. Furthermore, the CAR bound successfully to all represented subtypes of HLA-A2(the subtype specificity of the CAR itself was not stated),indicating that such a fine level of discrimination between donor and recipient haplotypes may not be achievable. MacDonald et al. [1]demonstrated that their HLA-A2-specific CAR-Tregs bound to HLA-A2 tetramers but not to control HLA-A24 tetramers;however,they did not test against individual subtypes of HLA-A2.In another CAR-Treg study, Boardman et al. [2] acknowledged that their choice of scFv against HLA-A2 was known to cross-react with HLA-A28 and HLA-68.
A paper by Tanigaki et al. [92] also noted that A2-binding peptides could potentially cross-react with other subtypes of the HLAA family—A24,A26,A28,and A29.Their results were derived from in vitro binding assays and do not demonstrate that CAR-Tregs will necessarily activate and exert suppressor functions upon binding.Nevertheless, to answer this question and improve the safety of CAR therapy, control testing of antigen specificity should ideally be performed across a panel of HLA molecules.
There is a positive role for cross-reactivity in the transplantation setting, however. As Boardman et al. [2] elucidated, crossreactivity allows for a number of different combinations of CAR specificity and the donor organ’s haplotype; therefore, this phenomenon can increase the number of patients who can benefit from receiving a particular CAR. Yet in order to exploit this possibility, knowledge of the full repertoire of each CAR’s binding targets is necessary to avoid detrimental binding to the recipient’s healthy tissue.
5.3. Presence of endogenous TCR specificity
Unless the TRAC gene is inhibited or knocked out,the CAR-Treg will retain its endogenous TCR with an independent specificity.This leads to early exhaustion if it comes into frequent contact with the TCR’s target antigen and may trigger the cell to act upon a nontarget tissue if aberrant binding occurs [122]. This concern has prompted efforts to replace endogenous TCRs with the aid of genome-editing technology such as CRISPR [131,132]. Eyquem et al. [122] experimented with a 2-in-1 approach of targeting a CAR to the TRAC locus itself in order to disrupt TCR expression in Tconvs, and reported an improvement in cell proliferation and effector function compared with control CARs with the TCR intact.Yet other studies suggest that functional dependency on the TCR varies between T cell subsets, and the findings of Eyquem et al.[122] may not be reproducible with CAR-Tregs. Tregs are reliant on TCR stimulation in order to maintain their suppressor function,and the expression of 25%of the activated Treg transcriptional signature is dependent on TCR signaling[133].This prompts the question of whether the signaling cascade induced by CAR activation can adequately mimic TCR-mediated activation in order to maintain the functionality of CAR-Tregs in the case of TCR ablation.Overall, there is insufficient data to recommend either inclusion or removal of endogenous TCRs.
5.4. Immunogenicity
Although autologous Tregs derived from the patient present no immunogenic concerns themselves, the presence of a transgenic CAR raises the potential for the detection of non-self proteins;this phenomenon has been previously recorded in a study of autologous Tconvs[134].Symptoms range from the formation of specific antibodies to acute side effects such as cytokine release syndrome(CRS).Therefore,reducing the immunogenicity of therapeutic cells is vital for reducing the risk to the patient, in addition to ensuring optimal survival and proliferation in vivo.
mAbs,the source of the scFv region,are principally derived from murine animals; this means that the variable chains of the CAR have the potential to provoke an anti-murine antibody response.Other proteins of non-human origin are also risk factors.Sommermeyer et al.[135]have demonstrated that the scFv region can instead be obtained from a human antibody chain library to avoid this risk,and the fusion sites within the CAR can be modified to produce a fully human construct.
5.5. CRS and neurotoxicity
While the predominantly anti-inflammatory cytokine repertoire of Tregs contradicts the pathogenesis of CRS and neurotoxicity,the frequency of these adverse events in CAR trials necessitates a discussion of this subject. CRS occurs when the activation of infused cells, combined with the subsequent activation of the host’s immune response, causes a surge in cytokine production. CRS is potentially life-threatening, leading to fever, vascular leakage,multiple organ failure,and even death[101].It is important to note that some key players in CRS, particularly IL-6, are secreted by endogenous monocytes and macrophages rather than by the CAR-expressing cells themselves [136]. Therefore, CRS remains a possibility despite the anti-inflammatory phenotype of CAR-Tregs.With this in mind, targeting the cytokine production pathways of endogenous cells may be a necessary therapeutic strategy where the genetic modification of CAR-expressing cells is insufficient to reduce the risk of adverse effects. Examples of such a strategy include the provision of the granulocyte–macrophage colonystimulating factor (GM-CSF) inhibitor lenzilumab in combination with CAR delivery, administration of the anti-IL-6Rα mAb tocilizumab, or pharmacologic blockade of catecholamine—although these options unfortunately add to the patient’s medication burden[137–141].
5.6. Phenotypic instability
The risk of adverse events can be exacerbated by any instability of the Treg phenotype, yet the stability of Tregs in vivo remains a contentious issue. Loss of FOXP3 expression and conversion to a proinflammatory state is a known phenomenon; Tregs have been reported to switch to a T helper 17(Th17)phenotype under certain inflammatory conditions,particularly when there is a deficiency of TGF-β in the microenvironment[28,142].On the other hand,many experts argue that fully differentiated, FOXP3highthymic Tregs are phenotypically robust,attributing the reported incidents of FOXP3 downregulation to the presence of non-committed developmental‘‘intermediates”and contamination with CD4+D25–cells displaying transient, promiscuous expression of FOXP3 [143–146]. While the question of whether true Tregs(and CAR-Tregs)are phenotypically stable in vivo is unlikely to be resolved in the near future,it is generally agreed that lineages of FOXP3highCD25highCD45RA–cells represent the most reliable population in terms of generational stability [143].
The epigenetic status of Tregs is a crucial factor for maintaining a stable lineage [143]. The conserved non-coding sequence 2(CNS2) within the FOXP3 locus must be demethylated in order for FOXP3 expression to be maintained after cell division, which is the case for thymic Tregs but not iTregs. Although it is possible to improve the functional stability of iTregs through ex vivo modification of their FOXP3 locus [147,148], instead, selecting thymic Treg populations through the processes of magnetic isolation or enrichment from cell samples has been the unanimous approach to CAR-Treg engineering for in vivo transplant studies to date, as well as in-human polyclonal Treg therapy [1–3,5,23,49,50].
It is also interesting to note that Nowak et al. [149] identified CD137+CD154–as a reliable signature for stable Treg phenotype after in vitro culture and reported that selecting for this signature,followed by further expansion of the purified phenotype, may increase the stability of the Treg population.
5.7. Tonic signaling
Tonic signaling—defined as chronic signaling through a receptor—can result in poor functional performance, cell exhaustion,and reduced persistence in vivo [150]. It can occur in either a ligand-dependent or ligand-independent manner, and may be instigated by activation of either the CAR or the native TCR. CD28 CARs exhibit a constitutive basal level of phosphorylation of the CD3ζ chain in their resting state, which renders them prone to exhaustion[95].Somewhat reassuringly,Noyan et al.[3]explicitly reported no tonic signaling within their HLA-A2-specific CD28 CAR-Tregs in their allogeneic transplant model,which would have been detectable through cell proliferation and signalling through the nuclear factor of activated T cells (NFAT) pathway without antigen stimulation. Nevertheless, further research is needed to support this result.The inclusion of a 4-1BB costimulatory domain has previously been reported to attenuate tonic signaling and hence reduce exhaustion in CAR-T cells [151]. However, others have counteracted this by showing that retroviral transduction of T cells with a 4-1BB CAR can lead to tonic signaling, reduced expansion, and impaired functionality [152]. Tonic signaling has been reduced by targeting the CAR sequence to the TRAC locus rather than to CD4, leading to greater persistence in vivo (which may be attributable to a decrease in cell activation events)[122,153]. Yet, as emphasized earlier in this review, disruption or downregulation of the TCR has consequences for the functionality of the Treg subset, so the CAR must be carefully targeted to avoid the loss of TRAC gene functionality.
5.8. Drug interactions
Achieving a tolerogenic environment exclusively with CARTregs is the ideal scenario for cellular therapy. It is, however,unclear at present whether an allogeneic response can be fully prevented without the aid of low-dose immunosuppressants.Consequently,it is important to identify the impacts of any pharmacologic drugs on the activity of the CAR-Tregs. Several categories of immunosuppressive agents act by targeting the IL-2 signaling pathway; these include basiliximab and calcineurin inhibitors[154–156]. Tregs are dependent on EC sources of IL-2 for survival and expansion,since(unlike Tconvs)they are incapable of producing IL-2 themselves, and there has been understandable concern over certain drugs potentially interfering with the Treg population in transplant recipients. Interestingly, multiple studies have reported that the short-term use of basiliximab, as per its role as an induction therapy, does not prevent the long-term persistence or the functionality of CD4+CD25+FOXP3+Tregs[155,157].
6. Considerations for commercial CAR-Treg production
6.1. Safety switches
The autonomy of therapeutic cells can lead to serious side effects that are difficult to control inside the patient. Given the largely undetermined dynamics of CAR-Tregs in vivo, it is important to include a safety mechanism by which these cells can be selectively deactivated or destroyed in the case of an adverse event during clinical trials and beyond.Published CAR-Treg studies have focused on immunosuppressive potential and have not yet explored safety strategies; however, these require careful consideration before the transition to in-human use.To allow for the external manipulation of infused CAR-Tregs,safety switches or suicide genes could be included either as part of the CAR or as a co-expressed molecule.The most extensively developed systems are based on genedirected enzyme prodrug therapy(GDEPT),inducible dimerization,and mAb depletion [158]. While the aforementioned methods are highly valuable in situations of serious adverse events, their effectiveness results in the irretrievable loss of therapeutic cells. This represents a loss not only of therapeutic benefit to the patient,but also of the time and costs associated with generating these cells.Alternatively, a reversible safety switch could provide a means of deactivating CAR-Tregs without destroying them. Research efforts are increasingly focusing on safety systems that do not result in instantaneous cell death, leading to innovations such as UniCAR technology and similar systems [159–161]. This is a better option in situations where destruction of the CAR+cells is not desirable.By stopping administration of the activating drug, CAR expression will be temporarily inactivated until re-administration.
From a transplantation perspective, one critique of these‘‘on-switch” systems is that they are disadvantageous for their dependency on the repeated administration of small-molecule drugs to maintain CAR activation. While this is acceptable for applications in which only short-term therapy is envisaged (e.g.,anti-cancer therapy), it is less suitable for transplant recipients requiring lifelong intervention of the immune system. This is also a reason why pharmacologic drugs are losing favor; the risk of non-adherence becomes more likely,and the costs associated with lifelong medication pose a heavy financial burden. An answer to this problem may lie in the tetracycline-responsive transcription(Tet-Off) system, whereby the gene of interest is expressed constitutively under the control of a tetracycline-controlled transactivator and tet operator sequences[162,163].Upon delivery of either tetracycline or doxycycline, the transactivator cannot bind to the operator sequences,and gene transcription is inhibited in a reversible manner [163]. Placing the CAR sequence under the control of a Tet-Off system would satisfy the need for a safety system while sparing the cells from irreversible destruction, and would present less of a burden to transplant patients.
6.2. Financial cost
At their current price range, the widespread application of CAR therapies could generate a financial strain on healthcare systems and might deter health insurance providers from covering the cost of infusion in countries without universal healthcare[164].At present, approved CAR therapies are offered only to a highly specific subset of patients suffering from relapsed or refractory disease,who have not responded to other treatments and who fulfil the age criteria [70]. In the context of transplantation, it is difficult to envisage how the cost will be managed if CAR-Tregs are approved as an immunosuppressive therapy.The‘‘last-resort”status of antitumor CAR-Tconvs is not applicable to transplantation, where it is hoped that Treg-mediated suppression will replace or partially substitute for pharmacologic drugs. While the benefits of HLAspecific, cell-based therapy are clear enough to envision that they will one day fulfil this role, the financial sustainability of lifelong immunosuppression is an important consideration.It is not certain how frequently CAR-Tregs will need to be administered. Largescale manufacturing of CAR products—autologous or allogeneic—may bring down the cost per infusion.
6.3. Optimizing dosage
One of the remaining questions regarding therapeutic application of CAR-Tregs is the quantity of cells required to achieve tolerance. Dosage varies greatly between studies and, at this point, the optimum number of cells per infusion remains debatable. It has been estimated from mouse models that a ratio of Tregs:Tconvs of at least 1:2 (33% Tregs out of the total CD4+T cells) is needed to prevent acute rejection of solid organ transplants [165]. This percentage of Tregs in the allograft cannot be maintained indefinitely,but having such a high proportion at the time of transplantation can establish a tolerogenic environment for a longer period through the mechanisms of bystander suppression and infectious tolerance [165]. Based on the assumption that an individual has 5 × 109CD4+T cells in the peripheral blood, with an average of 5%(2.5×108)of these being Tregs,an infusion of 4.9×1010Tregs would be required to achieve this ratio without lymphodepletion of Tconvs [165]. This would be a difficult number to achieve even with ex vivo expansion, so it can be argued that a combination of lymphodepletion and Treg infusion is the favorable strategy. With thymoglobulin lymphodepletion treatment,the Treg dose could be reduced to as low as 3 × 109–5 × 109cells [165].
However, the elevated potency of Tregs with predefined specificity may reduce the number of cells needed for infusion. CARTregs are capable of localizing preferentially to the allograft due to their scFv-derived binding site;therefore,the number of tolerogenic cells that infiltrate the graft will be increased. Further research is required to investigate this possibility, as well as the optimal frequency of CAR-Treg doses.
7. Summary and future perspectives
CARs are rapidly taking over the field of immunotherapy as their value as a potential tool for immune modulation gains recognition. The efficacy of CAR-Tregs in early murine studies—where they were shown to prevent symptoms of solid organ rejection to a greater extent than their polyclonal counterparts—is a strong argument that this technology is deserving of intense research efforts.Existing studies describing the generation of CAR-Tregs for the purpose of transplant tolerance have established the tolerogenic activity of CAR-Tregs and the antigen preference of the scFv, and have provided some information on persistence in vivo; nevertheless,other questions remain for long-term CAR-Treg therapy.
There is a fundamental need to expand the longevity of CARTregs in vivo. Data indicate that CARs bearing the commonly used CD28 costimulatory domain have sufficient suppressive capacity to prevent allograft rejection in vivo but exhibit reduced persistence,which may be attributed to a basal level of activation leading to earlier exhaustion, as previously discussed. It is necessary to explore combinations of other costimulatory domains and establish their effects on activation, proliferation, and tonic signaling in Tregs. Longer persistence means a greater timespan between infusions, which reduces the patient’s medication burden and the manufacturing costs associated with generating fresh batches of CAR-Tregs.
The major barrier that remains to be overcome is the unknown safety profile of HLA-specific CAR-Tregs in vivo. Despite the approval of two CAR-based therapies for cancer therapy by the FDA and the EMA, the outcomes of clinical trials have demonstrated several issues regarding the safety of CAR therapy.Although no clinical trials have taken place so far that could provide a report on the adverse effects of CAR-Tregs in humans,observations made through conventional CAR-T cell therapy may be used to create a repertoire of the potential side effects to be expected. Yet the unique functions of the Treg subset may pose a novel array of issues in the context of transplantation that have not yet been identified; for example, bystander suppression and the creation of a tolerant milieu may facilitate the survival of pathogens and malignant cells, causing long-term effects similar to those experienced under conventional immunosuppressive agents. A thorough examination of potential adverse effects, optimum dosage, and timing for adoptive transfer is therefore crucial before this field can progress beyond preclinical studies. If safety issues can be mitigated, and if further research is completed to improve our understanding of the behavior and dynamics of CAR-Tregs in vivo, then the therapeutic benefits of HLA-specific CARs are a reason for great optimism for their application in transplantation.
Acknowledgments
Fadi Issa is a Wellcome Trust Clinical Research Career Development(CRCD)Fellow.Work relevant to this review is supported by the European Union’s Horizon 2020 Research and Innovation Program (RESHAPE, 825392) to Joanna Hester and Fadi Issa.Sabrina Wright is supported by the Restore Research Trust.
Compliance with ethics guidelines
Sabrina Wright, Conor Hennessy, Joanna Hester, and Fadi Issa declare that they have no conflict of interest or financial conflicts to disclose.

- Engineering的其它文章
- Corrigendum to ‘‘Research on DC Protection Strategy in Multi-Terminal Hybrid HVDC System” [Engineering 7 (2021) 1064–1075]
- A Vision of Materials Genome Engineering in China
- Roles of Serum Amyloid A 1 Protein Isoforms in Rheumatoid Arthritis
- Ecological Barrier Deterioration Driven by Human Activities Poses Fatal Threats to Public Health due to Emerging Infectious Diseases
- Diverse Roles of Immune Cells in Transplant Rejection and Immune Tolerance
- Review on Drug Regulatory Science Promoting COVID-19 Vaccine Development in China