
抗菌肽多靶点作用抑菌机理研究进展
2022-06-06 01:44:06肖怀秋李玉珍林亲录赵谋明
食品与生物技术学报 2022年5期
肖怀秋,李玉珍,林亲录,赵谋明,刘 军,周 全
(1.湖南化工职业技术学院 制药与生物工程学院,湖南 株洲412000;2.中南林业科技大学 食品科学与工程学院,湖南 长沙410004;3.华南理工大学 食品科学与工程学院,广东 广州510641)
感染性疾病是由细菌和病毒等致病微生物感染引起的严重威胁公共健康安全的疾病,特别是以金黄色葡萄球菌为典型代表的多重耐药菌的出现使感染性疾病治疗变得更加复杂[1]。在抗生素替代药物研究中,抗菌肽(antimicrobial peptides,AMPs)因能使细菌无耐药性或降低细菌耐药性而备受关注[2],由基因编码的多肽前体通过蛋白酶解激活产生,大多数为阳离子(2~9价),一级结构中—NH2端常富含亲水性氨基酸,—COOH端富含疏水性氨基酸,疏水性残基一般超过30%,在细胞质膜中折叠后可形成疏水残基与亲水残基,分别位于两侧的双亲和空间结构,可形成α-螺旋和β-折叠等多种二级结构,具有广谱抗菌活性、热稳定性好、抗菌活性高、特异性强、有一定细胞选择性和对哺乳动物细胞毒副作用少等优势,对细菌、真菌和病毒甚至肿瘤细胞都有良好抑制活性[3],对生物体主动防御及主动免疫等也有重要作用,有望成为抗菌药物理想替代品[4]。抗菌肽对细胞具有多靶点单独或协同作用,研究抗菌肽多作用靶点抑菌机理对抗菌肽构效关系理论与应用研究具有重要指导意义。抗菌肽在生物体内的生理功能[5]见图1。

图1 抗菌肽生物学功能Fig.1 Biological functions of antimicrobial peptides
1 细胞壁损伤抑菌机制
细菌细胞壁主要含肽聚糖,真菌细胞壁主要含葡聚糖和甘露聚糖等成分。抗菌肽可通过与细胞壁合成前体分子结合(如脂质II分子带负电荷的焦磷酸糖[6-7])或干扰肽聚糖合成[8],或与肽聚糖和坦酸等细菌细胞壁关键组分结合[9]等方式造成细胞壁损伤。如β-defensin 3可与细胞壁合成前体焦磷酸糖结合,抑制细胞壁形成并破坏细胞正常形态,导致胞内容物外溢和细胞死亡[10];planosporicin通过阻断肽聚糖合成造成肽聚糖前体异常积聚,使细胞壁合成受阻[11];LL-37可作用于真菌细胞壁甘露聚糖造成细胞壁受损而表现抑菌活性[12];Teixobactin通过与肽聚糖前体Lipid II和坦酸前体Lipid III结合抑制细菌细胞壁合成而表现出抑菌活性[13]。
2 细胞膜损伤抑菌机制
细菌细胞膜上富含大量负电荷酸性磷脂,可通过静电吸附作用与阳离子抗菌肽结合,抗菌肽结构中疏水区域可与细胞膜两性离子磷脂表面聚集并富集在膜表面[14]。对G-细菌,抗菌肽可与负电荷细胞外膜脂多糖结合形成肽-脂复合物并形成跨膜通道破坏细胞膜完整性;对G+细菌,可与细胞表面坦酸结合并附着细胞表面,通过两亲性结构自聚形成构象簇穿过坦酸、脂坦酸、脂多糖及肽聚糖层等膜表面组分到达细胞膜并造成膜受损[15]。真菌细胞膜中麦角甾醇和鞘脂常为抗菌肽靶受体,如抗菌肽C16-Fengycin A通过抑制细胞膜中麦角固醇生物合成而破坏禾谷镰刀菌细胞膜结构[16],DmAMP1与细胞膜中鞘脂甘露糖苷二肌醇磷脂酰神经酰胺相互作用破坏酿酒酵母细胞膜结构[17]。阳离子抗菌肽不易与富含胆固醇及中性磷脂的哺乳动物细胞膜作用,因此,对哺乳动物细胞有一定细胞选择性[18]。抗菌肽细胞膜损伤模型主要有如下4种:
2.1 地毯(Carpet)模型
“地毯”模型是抗菌肽细胞膜损伤模型的典型代表。抗菌肽分子平行吸附于细胞膜表面脂双层,当抗菌肽浓度达到阈值后大量集聚在细胞膜表面形成“地毯”式结构,亲水端“伸向”水溶液,而疏水端通过静电吸附集聚于细胞膜表面,集聚浓度达到阈值后破坏细胞膜完整性,使细胞膜脂双层结构瓦解塌陷[19],抗菌肽其疏水核心并未嵌入到细胞膜内部,亲水区域也无须聚集形成孔道,当聚集到细胞膜表面的抗菌肽达到阈值后,细胞膜瞬间坍塌,细胞膜向内弯曲并引起膜破裂[19],作用于G-和寄生虫等抗菌肽常以毯式模型方式进行[20],如aurein、cathelidicins、indolicidin和LL-37等[21]。
2.2 环孔状(Toroidal-Pore)模型
抗菌肽分子极性端与细胞膜磷脂分子极性头部结合,插入到脂质双层膜并造成细胞聚集,细胞膜结构发生改变,亲水域形成孔内侧,而疏水域形成孔外侧,疏水性区域与磷脂分子头部基团协同形成“环孔”并在细胞膜上形成孔洞,造成脂质体与胞内生物大分子“外逸”。由于孔洞由抗菌肽与细胞膜脂质分子共同构成,微型抗菌肽也能形成“环孔”,抗菌肽以垂直方式插入细胞膜并使其疏水区发生移位导致细胞膜疏水中心形成裂口,诱导磷脂单分子层持续性向内弯曲形成跨膜孔道[22],该模型不存在特定肽-肽相互作用,肽与肽之间不干涉[23],最显著特征是双分子层排列,如magainin、LL-37和sticholysin II等[24-26]。
2.3 桶板(Barrel-Stave)模型
桶板模型的抗菌肽分子横跨细菌细胞膜形成具有中心内腔结构构象簇的跨膜离子通道,包括抗菌肽单体与细胞膜结合、单体间识别、连续多个单体嵌入与聚集、聚集抗菌肽单体以垂直方式排列细胞膜表面形成跨膜微孔4个步骤。其中,抗菌肽单体间识别与聚集是关键[24]。抗菌肽以束状形式垂直“覆盖”在细胞膜表面并不断往胞内渗透,在细胞膜上形成类似于木桶状的中空管腔结构,亲水部分朝向桶内壁构成亲水通道,疏水区域朝向膜内与磷脂结合,一个抗菌肽分子就相当木桶边沿上的“木板”,理论上需要至少4个“板”才能有效形成该模型结构。抗菌活性随抗菌肽分子数量增加而增加,抗菌肽分子越多内容物渗透就越严重[27],可破坏细胞正常渗透压并诱导细胞凋亡。Alamycin以α-螺旋构象附着、聚集并嵌入磷脂分子,其疏水性区域与磷脂分子疏水核心结合,亲水性区域形成跨膜离子通道或跨膜微孔[24]。桶板模型脂质疏水性和亲水性排列对细胞膜不会造成严重破坏,部分抗菌肽可易位到内质单层进入细胞质并与胞内组分发生相互作用[28]。只有少数抗菌肽可形成桶板模型,如alamethicin、pardaxin和protegrins等[3]。
毯式模型、环孔状模型和桶板模型抗菌肽对细胞膜的损伤机制见图2。

图2 抗菌肽破坏细胞膜作用机制Fig.2 Schematic representation of membrane-active mechanism of AMPs
2.4 “凝聚”(Aggregate)模型
细胞膜中磷脂先形成聚集体,当结合抗菌肽后聚集体形成的平衡态被打破并在细胞膜上出现离子泄露通道,促使胞内离子“逸出”并造成细胞凋亡。抗菌肽分子可竞争性取代脂多糖并与细胞表面Ca2+和Mg2+等金属阳离子结合而破坏细胞稳定性,也可与细胞内外膜上脂质层结合并破坏膜结构形成“孔洞”[3],与环孔模型相似,但抗菌肽排列没有特定方向,以聚集形式形成跨膜,只会增强细胞膜渗透性,不造成膜破裂,以无规则肽-脂质分子聚集形式插入细胞膜形成动态孔道[3],如maculatin1.1[29]和polyphemusin等[30]。
3 影响胞内生物大分子合成及代谢关键酶活性
陈旋等[31]研究发现,抗菌肽P7可抑制DNA复制和RNA合成导致大肠杆菌死亡;PR-39可通过抑制蛋白质与DNA合成以非裂解方式杀灭细菌[32];人源抗菌肽tPMP-1和aHNP-1进入到细胞后可抑制DNA与蛋白质合成[33];组蛋白衍生肽Buforin II在不改变膜通透性情况下通过细菌膜转运并直接与大肠杆菌胞内核酸或与组蛋白H2A结合,使DNA结构松散,削弱碱基堆积力,干扰DNA合成,BuforinⅡ突变体可与RNA相互作用表现更高结合活性,抑菌活性更强[34]。Indolicidin可阻止胸苷掺入到DNA复制过程,也可与细胞脂多糖结合抑制细胞壁合成,或抑制蛋白质生物合成[35]。Apidaecin可结合在细菌表面并转运进入到细胞质中,通过竞争性与细菌热休克蛋白结合,进而封闭并抑制伴侣蛋白质辅助众多未成熟蛋白质加工折叠,表现出良好的抗菌活性[36]。猪源抗菌肽PR-39可抑制胞内氧化酶复合物组装,导致细胞无法产生活性氧(ROS)并影响正常代谢[37]。部分α-螺旋肽(pleurocidin,dermasep-tin)和富含Pro与Arg的抗菌肽(PR-39,indolicidin)可阻碍(3H)胸腺嘧啶、(3H)尿嘧啶与(3H)亮氨酸在E.coli中表达,通过抑制核酸与蛋白质合成[32];富含Pro抗菌肽pyrrhocoricin、drosocin和apidaecin可抑制热激蛋白的ATP酶活性,影响蛋白质的正常空间结构形成[38]。Indolicidin能破坏细菌细胞膜且失活DNA拓扑异构酶来抑制DNA合成[39];Cathelicidin Bac7能靶向核糖体亚基并干扰蛋白质翻译,但不影响DNA复制和RNA转录[40];HNP-1和HNP-2能干扰细菌核酸与蛋白质合成并抑制周质β-半乳糖苷酶合成,破坏细胞壁完整性[41];Feglymycin(13肽)能抑制肽聚糖生成酶MurA和MurC活性,影响细胞壁形成[42]。
4 线粒体损伤抑菌机制
线粒体保持一定的膜电位对其正常生物功能发挥有重要作用,若线粒体受损或形态改变,则线粒体电位会下降,一旦膜电位耗尽,细胞凋亡则不可逆转,可作为线粒体早期损伤的重要表征指标,特别是线粒体介导的细胞凋亡。papiliocin[43]和psacotheasin[44]可改变线粒体电位耗散诱导白色念珠菌凋亡,CGA-N12和CGA-N9也可通过类似机制诱导热带念珠菌凋亡[45-46]。线粒体中Ca2+是真核细胞调控关键离子,当细胞过度兴奋或刺激时,线粒体对Ca2+的吸收会增强并诱导线粒体内膜损伤,使部分细胞凋亡因子(包括Cyt C)发生外溢[47]。核酸内切酶为Ca2+敏感酶,与细胞凋亡晚期染色质浓缩和DNA断裂密切相关,Ca2+超载诱导产生的ROS可直接作用于核酸并导致细胞核损伤与细胞凋亡[48]。Scolopendin可诱导线粒体吸收过量Ca2+并造成胞内氧自由基过量生成,从而诱导白色念珠菌因自由基过量而出现细胞凋亡[49]。CGA-N12和CGA-N9也可促进线粒体Ca2+吸收,诱导细胞内ROS过量产生并造成细胞凋亡[45-46]。促细胞凋亡因子Cyt C存在于线粒体膜空间,其释放常视为线粒体外膜受损或通透性改变,是真核细胞程序性死亡重要特征[50-51]。Melittin可增强线粒体对Ca2+的吸收,诱导胞内产生大量羟基自由基(·OH),并诱导细胞凋亡因子(Cyt C)泄漏,从而造成白色念珠菌线粒体和Yca1依赖性凋亡途径[52]。CGA-N12也可使热带念珠菌Cyt C泄漏,通过天冬氨酸蛋白水解酶依赖性途径诱导细胞凋亡[45]。线粒体负责TCA循环与氧化磷酸化等能量代谢途径,TCA循环可产生NADH和FADH2等辅酶,氧化磷酸化是利用辅酶生成能量并将质子逆浓度梯度泵入线粒体间隙,形成线粒体内外膜电化学梯度(电势用于ATP合成)。histatin 5可抑制辅酶I依耐性酶的生物活性(如苹果酸脱氢酶),抑制三羧酸循环并下调ATP合成酶的亚基(γ链),上调参与蛋白质生物合成的关键蛋白质表达水平,使胞内生物大分子泄漏,降低细胞的环境适应性并诱导细胞程序性凋亡[53]。LL-37可降低生物氧化呼吸链中相关基因的表达,使能量代谢出现障碍,从而表现抑菌活性[54]。常见抗菌肽结构氨基酸残基序列及抑菌作用机制见表1。
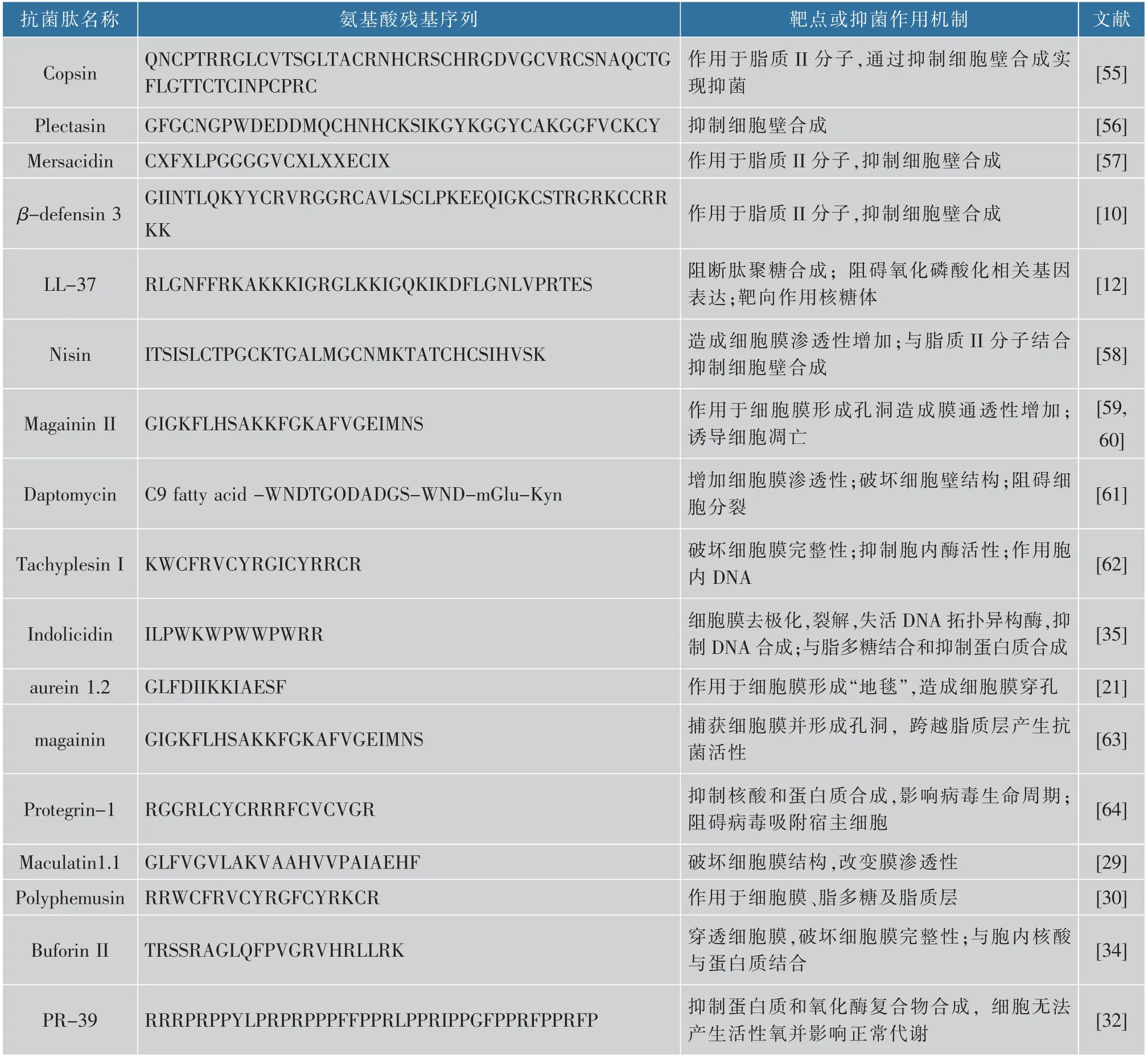
表1 常见抑菌肽结构氨基酸残基序列与靶点/作用模式Table 1 Sequence and target/action mode of amino acid residues of common AMPs

续表1
5 免疫调节抑菌机制
除直接杀死微生物外,抗菌肽在亚治疗剂量下可作为免疫效应分子调动并激活机体免疫细胞抑制或杀灭炎症[67]。LL-37可在机体感染或促炎因子诱导下,在生物体内组织或器官中表达和释放至黏膜表面发挥间接趋化作用[68],LL-37既可直接上调单核细胞趋化蛋白-1和IL-8表达,还可诱导单核细胞(monocyte)产生大量IL-1β,间接诱导白细胞介素和单核细胞趋化蛋白质表达,或上调趋化因子受体CXCR-4、CCR2和IL-8RB表达,招募免疫细胞到感染部位[69],或抑制丝裂原活化蛋白激酶(MAPKs)活化和Akt信号通路降低了促炎细胞因子分泌,减弱炎症反应[70],还可与细菌脂多糖(Lipopolysaccharide,LPS)结合降低内毒素介导的炎症反应[71];部分抗菌肽还可趋化免疫细胞至感染部位,如βdefensin hBD1和hBD2可结合单核细胞趋化因子受体并招募T细胞[72]来发挥抗感染作用。
6 展 望
细胞膜对维持细胞渗透压、控制物质交换、信号传导和能量生成等生化过程有重要作用,受损后会导致胞内容物外泄与胞外物质进入胞内,改变细胞渗透压,对细胞正常生理功能产生严重影响并诱导细胞凋亡。近年来,陆续报道了许多新型抗菌药物,但以细胞膜作为效应靶点居多,主要是因为以细菌细胞膜作为抗菌效应靶点可以解决现有抗生素难以彻底杀灭处于减速生长期与休眠期的细菌这一问题[73];细胞膜结构相对保守和作用于细胞膜的快速杀菌效应也使得以细胞膜作为效应靶点具有天然优势;此外,由于细菌细胞膜组分与真核生物细胞膜组分的差异,使得以细菌细胞膜作为效应靶点的新药物研发具有更好的细胞选择性和安全性,且不易产生耐药性[74]。但有研究表明,长期使用抗菌肽也可能存在耐药性[75]。笔者认为,尽管细菌细胞膜是抗菌肽筛选优秀的效应靶点,但也应考虑基于细胞壁损伤、线粒体损伤、胞内生物大分子合成抑制及代谢关键酶靶点的新型抗菌肽研发[3],从而为抗菌药物的研究提供更多可供选择的效应靶点。
猜你喜欢
现代畜牧科技(2021年9期)2021-10-13 06:38:40
中成药(2019年12期)2020-01-04 02:02:24
中成药(2018年7期)2018-08-04 06:04:18
浙江工业大学学报(2017年5期)2018-01-22 02:03:42
中成药(2017年12期)2018-01-19 02:06:31
广东农业科学(2017年5期)2017-08-29 10:37:54
中成药(2017年5期)2017-06-13 13:01:12
广东饲料(2016年5期)2016-12-01 03:43:21
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
特产研究(2014年4期)2014-04-10 12:54:12
