
常规抗结核化疗对肠道菌群的影响
2022-05-24 02:22:50张晨晨余美玲谭卫国巫株华魏文静
实用医学杂志 2022年6期
张晨晨 余美玲 谭卫国 巫株华 魏文静
1广东省结核病控制中心(广州 510630);2深圳市慢性病防治中心(广东 深圳 518020)
结核病不仅威胁人类健康还给全球经济造成巨大负担,2020年全球新发结核病患者987 万,发病率127/10 万,病死率15%[1]。针对药物敏感型结核病患者,世界卫生组织(WHO)推广使用标准短程化疗方案:利福平(RIF,R)、异烟肼(INH,H)、乙胺丁醇(EMB,E)和吡嗪酰胺(PZA,Z)联合使用2 个月,然后继续服用RIF 和INH 至少4 个月[2]。肠道菌群参与机体物质代谢和免疫应答反应,成为目前研究的热点之一[3-6],且对维持呼吸系统稳态,尤其是肺部的稳态发挥着重要作用[7-8],多种抗生素长期联合使用,可能会改变结核患者肠道菌群的多样性和结构,进而影响治疗结果。
与16s rDNA 高通量测序相比,宏基因组测序技术不仅检测速度快、灵敏度高,还可对样本中菌群多样性、种群结构、进化关系、功能活性以及相互关系等进行综合全面分析[9],本研究比较了健康人群、潜伏性结核感染(latent tuberculosis infection,LTBI)者、HRZE 四联抗生素抗结核化疗期间不同时间点结核患者肠道微生物群的变化,初步探索了结核分枝杆菌(Mtb)感染以及抗结核药物强化治疗对肠道菌群的影响,为抗结核药物合理使用及改善肠道菌群提供参考。
1 对象与方法
1.1 研究对象该研究为病例对照研究,依据《潜伏性结核感染管理指南》和《WS288-2017 肺结核诊断》,经临床、实验室及影像学检查方法筛选LTBI和结核病患者。选取2016年11月至2017年12月在深圳市慢性病防治中心进行体检的15 名健康者为Un组,其中女7例(46.67%),平均(34.80±10.35)岁。另选同一研究周期该中心确诊的γ 干扰素释放试验为阳性的16 例LTBI 者为LT 组,其中女5 例(31.25%),平均(38.00 ± 10.64)岁。17 例初诊肺结核且未进行治疗的患者为T0 组,其中女4 例(23.53%),平均(35.76±12.61)岁。T0 组经一线抗结核药物联合治疗2 个月后记为T2 组(17 例);T0 组经6 个月治疗且治愈后记为T6 组(17 例);T0组经6 个月治愈并完成2 个月随访者记为T8 组(7 例),其中因人口流动等原因有10 例未完成随访。各研究队列的性别和年龄比例差异均无统计学意义(P>0.05)。纳入标准:常住深圳人口,生活作息正常;不抽烟酗酒;无其他如心、肝、肺、肾功能障碍等重大疾病;无HIV 感染、急性恶性肿瘤和糖尿病史等;纳入研究队列前至少2 个月禁用微生态制剂和抗生素[10]。深圳市慢性病防治中心医学伦理委员会已审核通过本研究方案,且所有研究对象均已签署知情同意书。
1.2 粪便样品采集及微生物DNA 提取用无菌采集管留取各研究对象的新鲜粪便样本(样本量>1 g),低温条件下迅速转至实验室的-80 ℃超低温冰箱,可长期保存备用。样品DNA 提取可采用粪便DNA 微型试剂盒(MP Biomedicals)。
1.3 宏基因组测序及信息分析流程将合格的DNA 样品送至北京诺禾致源科技股份有限公司进行宏基因组测序,信息分析流程如下:(1)测序结果预处理:使用Readfq(V8)对Illumina HiSeq 测序平台的原始数据进行预处理,获得后续分析的有效数据,如果样品存在宿主污染,则采用Bowite2 软件(version2.2.4)过滤掉可能来源于宿主的reads。(2)Metagenome 组装:使用SOAP denovo软件(V2.04)对有效数据组装分析。(3)基因预测及丰度分析:采用MetaGeneMark 软件进行ORF(Open Reading Frame)预测及过滤,并用CD-HIT(V4.5.8)软件去冗余,再用Bowite2 软件获得后续分析的gene catalogue(Unigenes)。(4)物种注释:使用DIAMOND 软件(v0.9.9.110)将Unigenes 从NCBI 的NR 数据库(Version:2018-01-02)中抽提出的细菌、真菌、古生菌和病毒序列进行比对,运用LCA 算法进行物种注释。(5)常用功能数据库注释:将Unigenes 与功能数据库KEGG 数据库(Version 2018-01-01)、egg-NOG 数据库(Version 4.5)、CAZy 数据库(Version 201801)比对,选取丰度排名前34 的功能及它们在每个样品中的丰度信息绘制热图,并从功能差异层面进行聚类。
1.4 统计学方法使用GraphPad Prism 7 软件绘图,SPSS 17.0 软件分析数据,Kruskal-Wallis 检验用于比较分析各研究队列非冗余基因数(number of non-redundant genes)和多样性差异,当P<0.05为差异有统计学意义。
2 结果
2.1 测序结果概述本研究所得原始数据共计741 864.38 Mbp,平均数据量8 335.55 Mbp,质控所得有效数据736 782.68 Mbp(有效率99.32%),平均数据量8 278.46 Mbp。样品经单独及混合组装,所得Scaftigs 共计10 561 556 134 bp,平均长度2 218 bp。进一步的,基因预测由MetaGeneMark 软件完成,共得到12 877 969 个ORFs,平均每个样品143 089 个ORFs;经去冗余后,共获得2 629 714个ORFs,总长1 937.35 Mbp,平均长度736.72 bp,GC含量47.04%,其中完整基因个数为421 096,占所有非冗余基因总数的16.01%。使用blastp 将非冗余基因集比对到MicroNR 库,注释到属和门的比例分别为63.83%,87.22%。非冗余基因集的常用功能数据库注释可由DIAMOND 软件分析完成(e-value 不大于10-5),比对到KEGG 数据库、eggNOG 数据库和CAZy 数据库的ORFs 分别为1 752 149(66.63%)、1 714 019(65.18%)和84 677(3.22%)。
2.2 Mtb 感染对肠道菌群的影响
2.2.1 Mtb 感染后肠道菌群菌群多样性变化从基因在各样品中的丰度表出发,随机从Un、LT 和T0 各研究队列中分别抽取不同数目的样品,将得到的不同样品组合间的基因数目用于绘制Pan 基因稀释曲线,见图1A。与Un 组和LT 组相比,T0组肠道菌群中的非冗余基因数未发生显著变化(均P>0.05,图1B)。利用Alpha 多样性分析法对不同组别间菌群丰富度进行分析,计算Shannon 指数,统计分析发现Un、LT 和T0 三组研究对象肠道菌群丰富度指数差异无统计学意义(均P>0.05,图1C),说明Mtb 感染未引起人肠道菌群丰富度的改变。Beta 多样性反应了各样本间菌群的整体差异程度,两样本之间的距离越大表示两者间的群落构成差异程度越大,基于Bray-Curtis 距离指标,本研究使用主坐标分析法(principal co-ordinates analysis,PCoA)进一步分析了Mtb 感染对各组肠道菌群结构多样性的影响,T0 组与Un 组在PC2 上存在显著的菌群结构差异(P=0.013 4,图1D)。
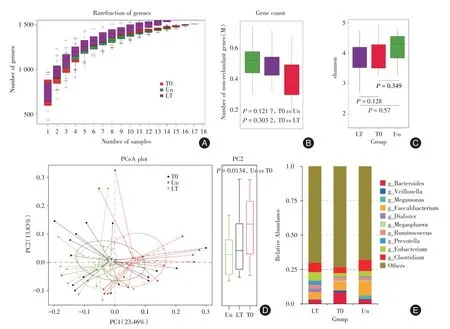
图1 Mtb 感染后肠道菌群多样性变化Fig.1 Changes of gut microbiota diversity after Mtb infection
2.2.2 Mtb 感染后肠道菌群结构组成本研究统计了Un、LT 和T0 组在属水平上各微生物的相对丰度(图1E),结果显示,三组中物种丰度较高的属有10 种,T0 组中梭菌属(g__Clostridium)的相对丰度为4.28%,较Un组(8.09%)和LT组(6.80%)减少;LT 组和T0 组中巨单胞菌属(g__Megamonas)的相对丰度分别为0.26%和0.16%,比Un 组(2.58%)明显减少;LT 组中优杆菌属(g__Eubacterium)、普雷沃菌属(g__Prevotella)和小类杆菌属(g__Dialister)的相对丰度分别为6.27%、3.01%和2.60%,比Un组(3.98%、0.57%和0.09%)和T0 组(2.74%、0.39%和0.07%)增加;T0 组中韦永氏球菌属(g__Veillonella)和拟杆菌属(g__Bacteroides)的相对丰度分别为1.54%和8.28%,较Un 组(0.32%、3.32%)和LT 组(0.21%、3.07%)明显增加。
2.3 常规抗结核化疗对结核病患者肠道菌群影响
2.3.1 常规抗结核化疗对结核病患者肠道菌群多样性影响随机从T0、T2、T6 和T8 各研究队列中分别抽取不同数目的样品,将得到不同样品组合间的基因数目用于绘制Pan 基因稀释曲线,如图2A 所示。分别与T2 组、T6 组和T8 组进行比较,T0 组患者肠道菌群中的非冗余基因数差异有统计学意义(均P<0.05,图2B)。Alpha 多样性分析发现,T0 组、T2 组和T6 组间的Shannon 指数差异存在统计学意义(P=0.008 8,图2C),说明结核病患者经常规抗结核化疗后,肠道内微生物的丰富度明显降低。基于Bray-Curtis 距离指标,使用主坐标分析法(PCoA)分析常规抗结核化疗对结核病患者肠道菌群结构多样性的影响,见图2D,T0 组与T6 组在PC1 上存在显著的菌群结构差异(P=0.002 1)。
2.3.2 常规抗结核化疗对结核病患者肠道菌群结构组成影响统计T0、T2、T6 和T8 组在属水平上各微生物的相对丰度(图2E),结果显示,三组中物种丰度较高的属有10 种,相对丰度最高的为拟杆菌属(g__Bacteroides);随着常规抗结核化疗时间延长,韦永氏球菌属(g__Veillonella)、小类杆菌属(g__Dialister)和普雷沃菌属(g__Prevotella)在T0组(1.54%,0.07%,0.39%)、T2 组(3.88%,2.46%,1.37%)、T6(6.89%,2.91%,1.72%)组和T8组(7.79%,3.24%,2.65%)中的相对丰度逐渐增加;梭菌属(g__Clostridium)和粪杆菌属(g__Faecalibacterium)在T0 组(4.28%,6.10%)、T2 组(3.07%,8.47%)、T6组(3.00%,4.25%)和T8 组(2.21%,1.41%)中的相对丰度逐渐减少。
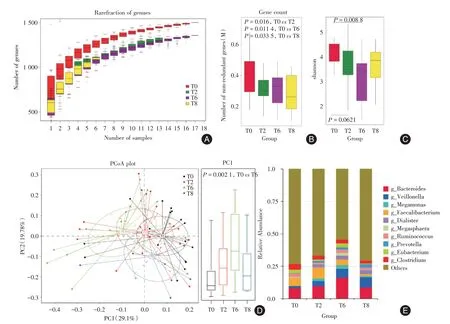
图2 常规抗结核化疗对结核病患者肠道菌群多样性影响Fig.2 Effects of conventional anti-tuberculosis chemotherapy on gut microbiota diversity of tuberculosis patients
2.4 差异菌群分析在属水平上,选取Un、LT、T0、T2、T6 和T8 组样本中相对丰度排名前35 位的物种进行分析,用热图进行可视化展示寻找变化差异较大的菌种,并从物种层面进行聚类,从而找出各组中聚类较多的物种(图3)。T0 组中的Erysipelatoclostridium和解黄酮菌属(Flavonifractor)与Un 组相比,显著增加;而梭菌属(Clostridium)和嗜血杆菌属(Haemophilus)较Un 组则显著减少。T8组中支原体属(Mycoplasma)、优杆菌属(Eubacterium)、Dorea、扣林氏菌属(Collinsella)、颤杆菌属(Oscillibacter)、芽殖菌属(Gemmiger)和粪杆菌属(Faecalibacterium)较T0 组中显著减少。为进一步发现高维度生物标识,通过线性判别分析效应量(linear discriminant analysis effect size,LEfSe)展示了各组在不同分类水平上有显著性差异的物种(图4),柱状图的长度(即LDA SCORE,默认设置为4.0)代表了差异物种的影响大小。
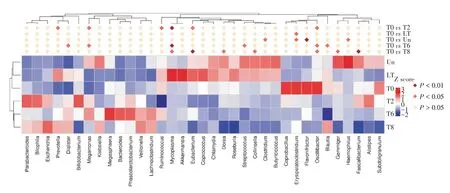
图3 属水平上差异物种的热图Fig.3 Heat map of different species at the genus level
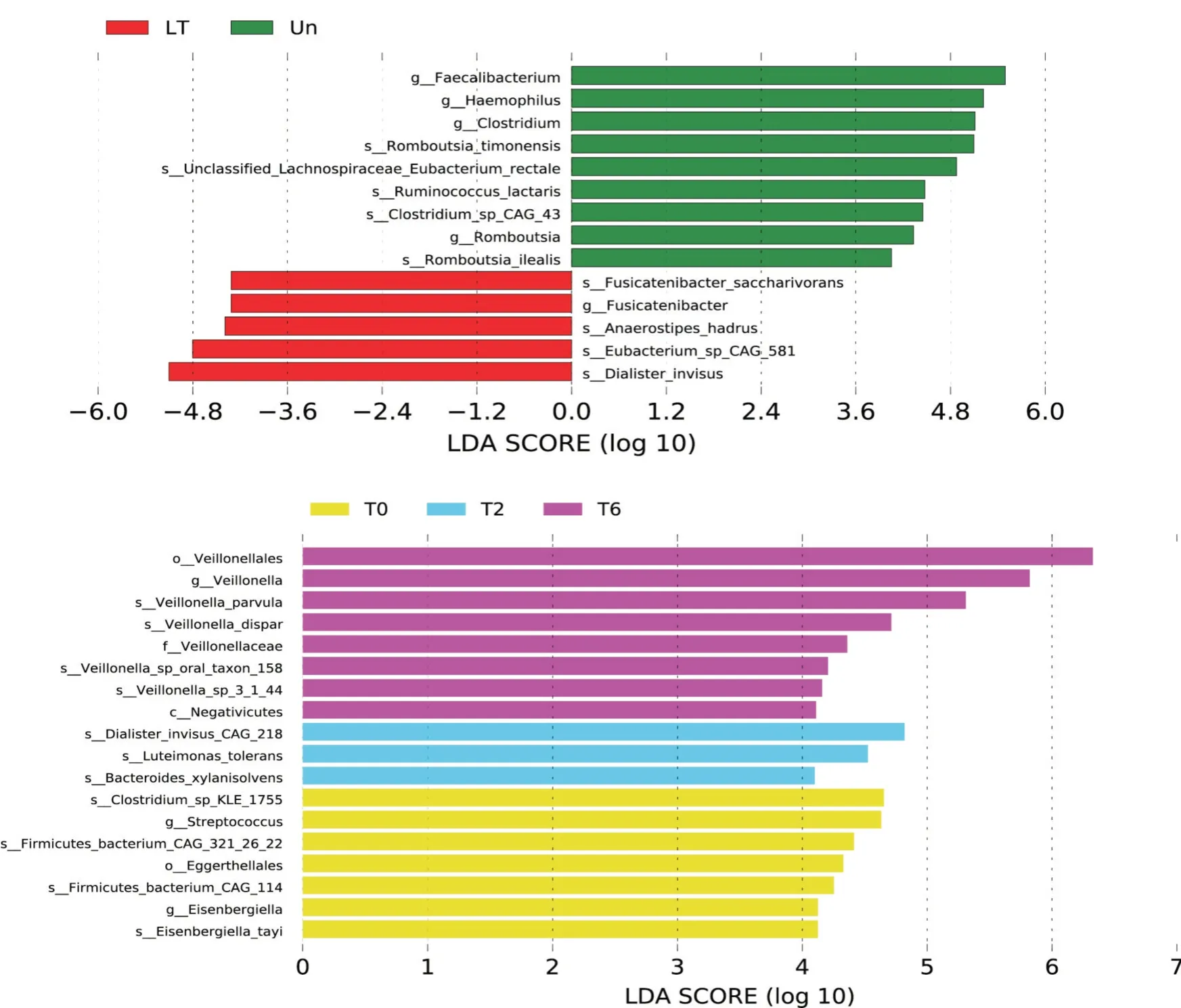
图4 LDA 值分布柱状图Fig.4 Bar chart of LDA score distribution
2.5 基于物种丰度的降维分析用无度量多维标定法(NMDS,Non-Metric Multi-Dimensional Scaling)分析Un、LT、T0、T2、T6 和T8 组样本的组内或组间差异,图中各点之间的相互距离表示不同样本间的差异大小。见图5,Stress=0.132,表明NMDS 分析是可信的,在MDS1 上,各组间比较差异无统计学意义,表明各组样本的物种组成无明显变化;而MDS2 上,T6 组与Un、LT、T0 和T2 组相比,菌落组成差异较大(均P<0.05)。
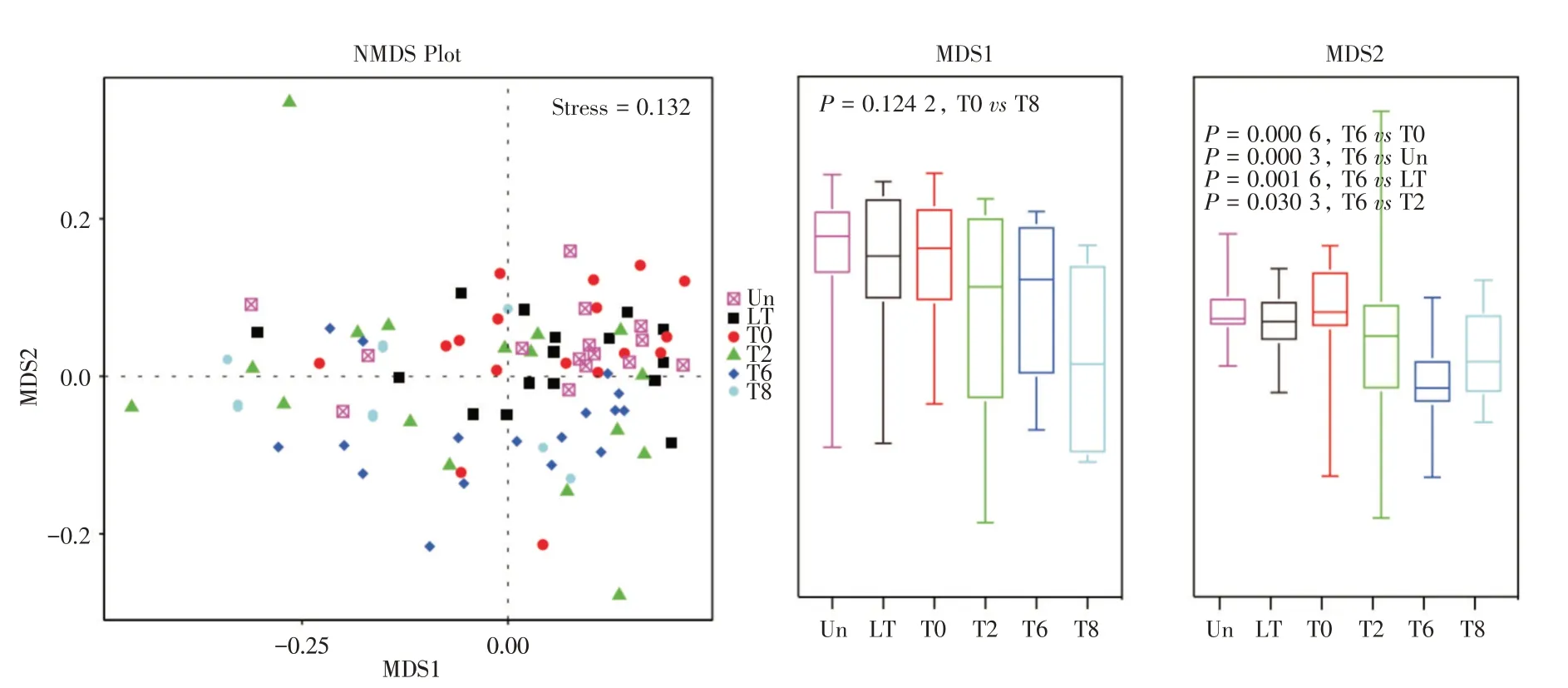
图5 属水平上的NMDS 图Fig.5 NMDS diagram at the genus level
2.6 基因功能注释分析见图6,与T0 组相比,结核病患者经常规抗结核化疗后,肠道内微生物群中部分功能途径发生变化,其中最显著的变化是抗结核治疗过程中辅助因子和维生素代谢(Cofactor and vitamin metabolism)、脂多糖代谢(Lipopolysaccharide metabolism)、乙醛酸和二羧酸代谢(Glyoxylate and dicarboxylate metabolism)、原核生物的碳固定途径(Carbon fixation pathways in prokaryotes)和柠檬酸循环(citrate cycle)功能显著升高。

图6 功能丰度聚类热图Fig.6 Clustering heat map of functional abundance
3 讨论
肠道菌群失调可导致炎症反应,通过肠-肺轴降低肺部对外来病原体的清除能力,所以肠道菌群的稳态对肺部健康起着重要作用[11]。为有效预防和治疗结核病提供理论基础,本研究选取了结核分枝杆菌感染及抗结核化疗不同阶段的患者样本,通过宏基因组测序分析发现,结核病患者与健康人群、LTBI 者相比,肠道菌群中非冗余基因数和Alpha 多样性均无明显变化,而菌群结构组成则存在较大差异。结核病患者肠道中梭菌属的相对丰度降低,而韦永氏球菌属和拟杆菌属的相对丰度则明显增加。近年来,有大量研究[12-14]结果显示,结核分枝杆菌侵入机体后导致其肠道菌群结构发生改变。OSEI 等[15]的研究显示,结核分枝杆菌侵染小鼠导致其肠道内厚壁菌门和梭菌目的数量明显减低;相类似的,结核分枝杆菌侵入人类和恒河猴后,未明显改变其肠道中微生物数量、丰富度和多样性,但却导致其肠道中脆弱类杆菌(Bacteroides frafilis)等类杆菌的数量明显增加,而厚壁菌门和梭菌目的数量明显降低[16],这与本文的研究结果相一致。
与初诊结核病且未进行治疗的肺结核患者相比,常规抗结核化疗后结核病患者肠道菌群中的非冗余基因数、Alpha 多样性和菌群结构都出现显著性差异。近年来,细菌16S rDNA 测序和宏基因组测序技术普遍应用于人体肠道菌群结构及功能改变的研究中,并获得了大量的数据[17]。HU 等[16]发现,结核分枝杆菌侵入机体会略微降低其肠道微生物多样性,并未对肠道菌群结构产生显著变化,但经常规抗结核化疗后其菌群多样性和结构发生明显改变,其中拟杆菌属相对丰度升高,而粪杆菌属和活泼瘤胃球菌的相对丰度则明显降低,这与本文的研究结果相似。而WIPPERMAN 等[18]对正在进行常规抗结核化疗和已经治愈的结核病患者进行对比研究发现,两组患者的肠道菌群多样性改变并不明显,但在随访的1~2年内其菌群结构则存在明显改变。不同的研究设计,受到地域、生活习惯等复杂外源性因素和遗传背景影响[19],所得结论难以统一,但基本可以确定抗结核化疗可导致肠道菌群结构发生明显变化,且可持续至停药后较长时间。
抗结核药物虽对结核病治疗有明显疗效,但对肠道的物质代谢却造成了严重影响。MAJI 等[20]的研究表明,肺结核患者进行常规抗结核化疗后,其肠道微生物中与代谢相关的功能基因出现改变,其中与氨基酸及辅酶合成和维生素代谢相关基因降低,丙酸盐和丁酸盐代谢通路基因升高,致病性相关基因升高。另有研究[18]表明,因肠道菌群结构改变导致其物质代谢也发生相应改变,如胆汁酸合成量降低,而脂肪酸氧化及维生素合成升高等。不同的研究[21]中,肠道菌群功能变化有差异,这可能与样本量的多少、肠道菌群的复杂程度、测序及分析方法等多种因素有关。本研究通过功能注释,发现抗结核化疗过程中辅助因子和维生素代谢及脂多糖代谢功能显著升高。
综上所述,与机体感染结核分枝杆菌相比,抗结核化疗造成了肠道菌群多样性、结构组成和代谢功能等多方面的改变。了解肺结核患者肠道微生物改变对机体免疫调控和开发新的分子标志物都具有推动作用;另外,未来在抗结核化疗过程中,重建患者的肠道微生物群有望提高结核病的治疗效果。本研究对同一结核病患者的不同抗结核化疗阶段进行了追踪随访,研究结果有较强的参考价值,但每组纳入的研究对象数量相对较少,后续还需扩大各研究队列的样本量、优化随访方法并延长随访时间,进一步深入的探索结核分枝杆菌感染及抗结核化疗对肠道菌群的影响。
猜你喜欢
西部医学(2024年3期)2024-03-21 12:22:24
中老年保健(2022年2期)2022-08-24 03:20:50
保健医苑(2022年5期)2022-06-10 07:46:46
昆明医科大学学报(2021年4期)2021-07-23 01:22:00
科学(2020年4期)2020-11-26 08:27:06
中国民族医药杂志(2016年4期)2016-05-09 07:41:11
中国医药生物技术(2015年4期)2015-12-26 08:26:36
动物营养学报(2015年10期)2015-12-01 02:26:20
中国卫生(2015年1期)2015-11-16 01:06:02
结核与肺部疾病杂志(2015年4期)2015-07-18 11:08:22
