
原发性肺腺样囊性癌及黏液表皮样癌的基因突变及表达谱系构建
2022-05-20 08:44:36吴桢珍
南京医科大学学报(自然科学版) 2022年5期
韩 玉,何 薇,吴桢珍,李 翀,丁 浩,赵 欣*,黄 茂*
1南京医科大学第一附属医院呼吸与危重症医学科,江苏 南京 210029;2常州市第一人民医院呼吸内科,江苏 常州 213000;3镇江市第一人民医院呼吸内科,江苏 镇江 212000
在2015年版WHO 肺肿瘤分类中,肺涎腺肿瘤包括腺样囊性癌(adenoid cystic carcinoma,ACC)、黏液表皮样癌(mucoepidermoid carcinoma,MEC)、上皮肌上皮癌、多形性腺瘤[1],以前两者最常见。肺涎腺肿瘤生长常较慢,对于病灶局限的肿瘤组织,手术切除是首要治疗方式[2],但对于无法手术的患者,想要探究更多的治疗方案,需要对肺涎腺肿瘤的发生发展机制有更深的研究,目前对肺涎腺肿瘤的报道主要包括一般情况、临床特征、预后,且多为个案报道和临床分析,在疾病发生、转移等机制方面的研究较少。高通量测序,又称下一代测序(next genera⁃tion sequencing,NGS)能够准确检测被检测组织所有的基因结果,且因其能快速、准确地检测而被广泛用于肿瘤患者的基因检测,具有检测量大、精确度大的优势。本研究通过NGS 检测技术从DNA 及RNA两个层面对肺ACC、肺MEC进行全面分析。
1 材料和方法
1.1 材料
研究纳入2017 年5 月—2020 年7 月南京医科大学第一附属医院、常州市第一人民医院、镇江市第一人民医院共计39 例患者,其中,ACC 14 例,MEC 24 例,1例患者因病理不清在后续基因检测过程中被剔除。本研究经南京医科大学第一附属医院伦理委员会批准(伦理审批号:2022⁃SR⁃254),患者经手术、支气管镜及经皮肺穿刺获取组织送检病理前均已签署知情同意书,收集患者的肿瘤组织样本及可获取到的部分癌旁样本用于后续分析。所有病例的肺涎腺肿瘤均为原发。
1.2 方法
1.2.1 组织基因检测
采用QIAGEN DNA 和QIAGEN RNA 提取试剂盒进行组织样本DNA 及RNA 的提取,实验操作流程参照提取试剂盒说明书。分别对提取后的DNA和RNA 进行Qubit 定量,其中DNA 提取总量不低于200 ng,RNA提取总量不低于200 μg。
DNA 建库富集:提取好的DNA 样本经超声打断,末端修复、接头连接、纯化基因后Qubit 定量,选取片段化处理后平均大小250~300 bp 的片段DNA进入测序流程;对选择的片段进行PCR 扩增及纯化、文库定量及PCR 扩增效率计算构建测序文库。DNA 富集选择南京世和基因生物技术股份有限公司自研的416 panel 进行肿瘤相关基因富集。将基因捕获探针与文库杂交,加入引物、杂交溶液、探针,捕获的文库与链霉亲和素磁珠结合,去除非特异性结合的文库。
RNA建库:提取的RNA通过KAPA建库试剂盒进行处理,首先通过RiboErase 去除核糖体核糖核酸(ribosome ribonucleic acid,rRNA),进一步去除RNA 样本中的DNA 残留,利用纯化的RNA 进行互补脱氧核糖核酸(complementary deoxyribonucleic ac⁃id,cDNA)合成,进一步合成互补链,末端修复,接头连接和文库扩增用于后续上机测序。RNA 部分检测无富集流程。
上机测序:按照Hiseq 4000 使用说明准备测序试剂,将表面标记的流式细胞上机测序;选用paired⁃end程序(基因片段采用桥式扩增法,而未经过环化的方式)进行双端测序,测序过程由Illumina提供的数据收集软件进行控制,并进行实时数据分析取得原始测序序列,使用Q30(原始数据中测序碱基质量数值大于30的碱基数量占总碱基数量的百分比)标准去除达不到标准的碱基;去除PCR扩增过程中产生的重复数据。
1.2.2 生物信息学分析
DNA 生物信息学分析:测序下机数据为bcl 格式,使用bcf2fastq(v2.17.1.14,Illumina 公司)软件将其转换为FASTQ(版本:Illumina 1.8+)格式。通过Trimmomatic软件进行原始测序数据质控,去除低质量reads(高通量测序仪产生的测序数据);通过BWA 软件(https://github.com/lh3/bwa/tree/master/bwakit)将得到的DNA 序列与人源基因组序列hg19比对。使用基因组分析工具包GATK 3.4.0(https://software.broadinstitute.org/gatk/)进行插入缺失突变和碱基质量进行评分,用VarScan2 检测体细胞突变。体细胞突变保留标准:对于在COSMIC 数据库中报道超过20 次的基因突变,最小突变丰度阈值(VAF)=1.0%,且至少有3根突变reads支持,对于其他突变VAF=2.0%,且至少有5 根突变reads 支持。此外,所有突变还需要满足最小测序深度≥20,最小碱基质量≥25,reads 偏倚不大于10%的标准。采用ANNOVAR 对单核苷酸多态性(SNP)和Indel(插入/缺失)注释,数据库:dbSNP(单核苷酸多态性数据库)(v137)、1 000Genome(千人基因组数据库)、Ex⁃AC(外显子组整合数据库)、COSMIC(癌症中体细胞突变目录)(v70)、ClinVAR和SIFT(尺度不变特征转换)数据库。1000Genome 或ExAC 项目中群体频率大于1%的突变被删除。进一步通过内部同一测序平台收集的随机测序错误数据库对得到的突变结果进行过滤,该列表由1 000 个正常样本的测序结果汇总而成,最小平均测序深度为700 x。举例来说,如果1 个组织样本突变(突变丰度2%reads 数5根)在内部随机测序错误数据库中的发生频率大于等于20%,将被认为是1 个随机错误被删除。拷贝数分析:通过对同批次上机对照样本进行主成分分析,降低拷贝数结果的系统噪音,以同批次同panel对照样本为基线,分析组织样本的拷贝数变异,缺失的阈值为0.65,扩增的阈值为1.5。
RNA 生物信息学分析:测序下机数据为bcl 格式,使用bcf2fastq 软件将其转换为FASTQ 格式。通过Trimmomatic 软件进行原始测序数据质控,去除低质量reads。融合变异分析:使用Bowtie aligner 进行转录组和基因组比对,再使用STAR(版本2.5.2b)进一步分析未比对成功的reads,最后采用Fusion⁃Catcher(version 0.99.4e)对上述参数进行融合变异分析。利用DESeq2(version 1.16.1)和edgeR(ver⁃sion 3.18.1)进行差异表达分析。差异表达分析中基因的差异倍数(fold change)>2,且P值<0.05。对应的火山图和热图分析等均基于本地的R代码分析。
2 结果
研究共计纳入39例ACC及MEC患者,其中1例患者因病理不清在后续基因检测过程中被剔除,38例患者中ACC 患者14 例,MEC 患者24 例。DNA 层面:对38例患者的肿瘤组织样本进行了416panel检测,DNA中位提取总量为1 645 ng(201~25 400 ng),关键测序质控原始测序深度875x,去重后测序深度531x,重复率33.3%;RNA 层面:38 例患者中1 例因RNA提取量不足未纳入后续分析,37例患者的所有肿瘤样本及15例患者的癌旁样本接受了RNAseq检测,RNA 中位提取总量为4 454 ng(276.1~27 064.0 ng),关键测序质控原始中位reads 数34 Mb,高质量reads(Q30)占比94.4%,比对率97.9%,DNA 中位污染率为5%。
分别从DNA和RNA层面对ACC及MEC肿瘤组织进行了突变层面和表达层面的分析,并综合了DNA层面突变及RNA层面融合分析结果。
ACC中发现8例患者携带MYB融合突变(57.1%),3例患者携带MYBL1融合突变(21.43%),融合伴侣均为NFIB基因。人的MYB基因是定位于6q22⁃23的原癌基因,通过表达c⁃MYB转录因子促进MYB蛋白合成来调节细胞的增殖与分化[3],既往研究发现MYB变异除6q22⁃23,还可发生于6q24、12q 和14q[4],本研究中MYB 断裂区域为外显子(exon)8/9/12/14,NFIB断裂区域为exon9/11。MYBL1基因是位于8号染色体的MYB基因的一种,主要激活c⁃MYB转录因子,既往在乳腺ACC中研究发现MYBL1也可通过与其他基因融合出现MYBL1过表达[5]。
原发性肺MEC中检测到12例患者发生MAML2基因融合(50%),具体形式为CRTC1⁃MAML2 融合突变。MAML2基因重排最早见于涎腺黏液腺癌,常见11号染色体长臂和19号染色体短臂的基因转位t(11;19)(q21;p13)[6],后在肺MEC 中也发现了该基因重排。本研究还检测到3 例EWSR1(12.5%)和1例ROS1(4.2%)融合患者,既往未在MEC 中提及。尤文肉瘤断裂点区域1(Ewing sarcoma beakpoint re⁃gion 1,EWSR1)因在尤文肉瘤中被发现而命名,位于22q12。ROS1 基因重排在本研究中融合形式为CD74:exon6~ROS1:exon33。
在肺ACC 及MEC 中分别检测到2 例及1 例EP300的错义突变。均检测到的突变基因还有AR⁃ID1A,ACC 中1 例患者检测到,MEC 中3 例患者检测到。
除DNA 检测,研究进一步从RNA 表达层面,通过对比肿瘤和癌旁样本分析了ACC 和MEC 的差异表达基因。使用DESeq2 进行基因差异表达分析并形成基因差异火山图(图1、2)。
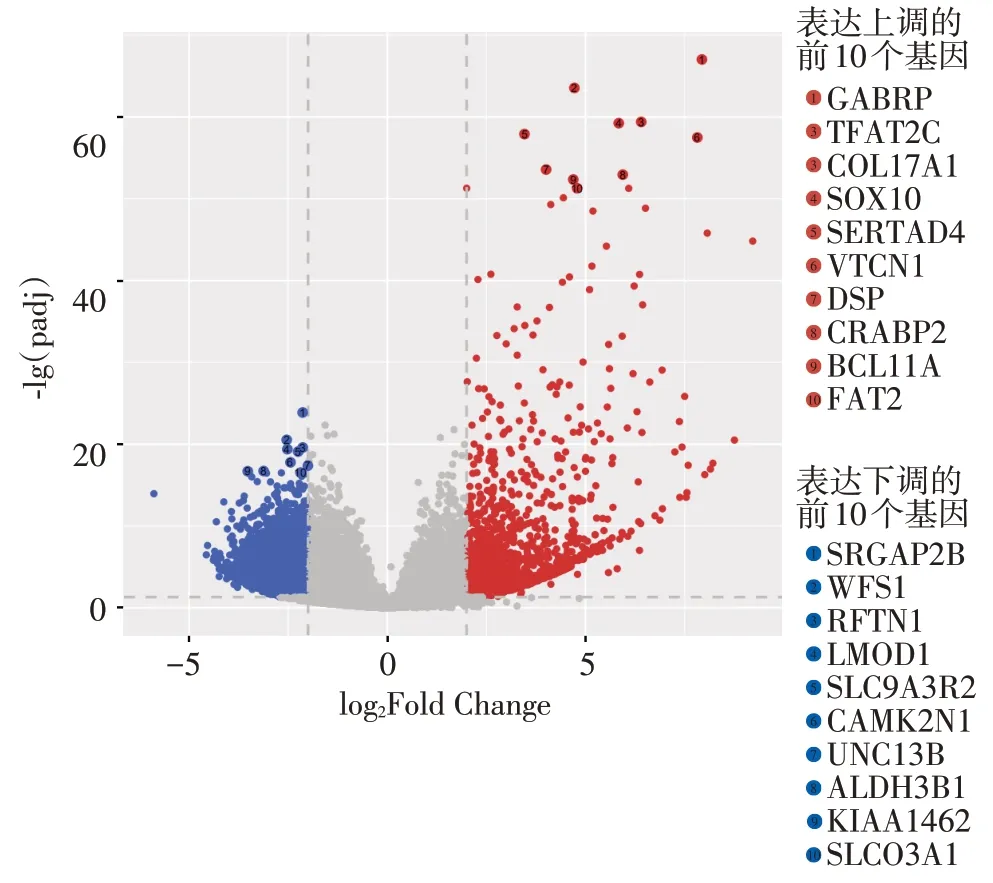
图1 肺ACC癌组织与癌旁组织差异表达的RNA所制的火山图Figure 1 Volcano plot of RNA differentially expressed in cancer tissue and adjacent tissue of lung ACC
肺ACC 差异表达结果显示表达上调的基因有GABRP、TFAP2C、COL17A1、SOX10、SERTAD4、VTCN1、DSP、CRABP2、VTCN1、DSP、CRABP2、BCL11A、FAT2,表达下调的基因有SRGAP2B、WFS1、RFTN1、LMOD1、SLC9A3R2、CAMK2N1、UNC138、ALDH381、KIAA1462、SLCO3A1。结合既往ACC 的研究,SOX10 及BCL11A 可能成为肺ACC的生物标志物。
肺MEC 差异表达结果显示表达上调的基因有TFAP2A、TRIM29、TNS4、EYA2、SERPINB5、FAM83F、KRT6A、FAT2、PPAP2C、PITX1,表达下调的基因有LIMCH1、EMP2、SPTBN1、HIGD1B、RAB11FIP1、ACVRL1、SHROOM4、CAV1、PTPRB、LDB1。表达差别第一的转录因子活化蛋白2A(TFAP2A)表达上调时,通过PDGFR、TGF⁃β和VEGFR 等信号通路促进肿瘤血管的生长、降低安罗替尼的抗血管活性,耐药发生率增加[7]。
3 讨论
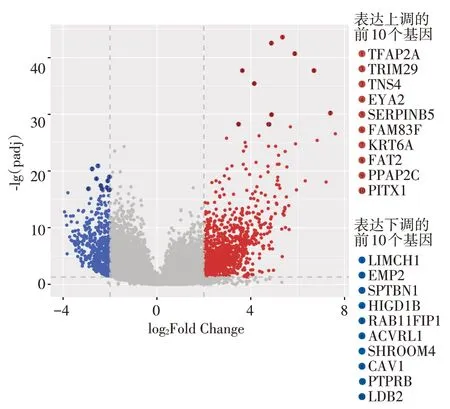
图2 肺MEC 癌组织与癌旁组织差异表达的RNA 所制的火山图Figure 2 Volcano plot of RNA differentially expressed in cancer tissue and adjacent tissue of lung MEC
肺涎腺肿瘤是肺部少见肿瘤,其中肺ACC主要发生在气管和支气管,肿瘤组织在镜下可见实性、管状、筛状结构,可见黏液样及透明样物质,细胞形态相对单一[8];肺MEC相较肺ACC更好发于叶支气管,由中间型细胞及表皮样细胞组成,形成大小不等的囊腔,细胞形态和组织结构多样[9]。既往研究提示,肺MEC在原发性肺癌中的比例低于1%[10],肺ACC 发病率为原发性肺肿瘤的0.1%~0.5%[2],因此目前原发性肺涎腺肿瘤的研究多集中在个案报道及临床资料总结,对基因突变的研究多为单个突变基因研究。本研究检测了共38 例原发性肺ACC 及肺MEC 肿瘤组织及癌旁组织的突变DNA 及与癌旁组织表达差异的RNA,首次对多例原发性肺MEC及ACC 的DNA 及RNA 反转录组测序并分析,而基因图谱的分析对靶向治疗的选择具有重要意义。
结合本研究及既往研究结果,肺ACC中最常见突变为MYB/MYBL1融合,与既往研究相符,MYB基因在白血病、结肠癌、乳腺癌、黑色素瘤中有较多研究;通过形成MYB⁃NFIB融合参与肿瘤的发生发展,NFIB 位于9 号染色体,周梦娇等[11]研究发现NFIB可作为增强子与MYB启动子结合促进MYB蛋白表达,参与细胞增殖分化。既往对唾液腺腺样囊性癌的研究提示t(6;9)(q22⁃23;p23⁃24)为特征性MYB⁃NFIB融合[12],头颈部唾液腺腺样囊性癌中该种融合比例可达16%~100%[13],目前对ACC 中MYB⁃NFIB融合的研究多来自唾液腺ACC,肺ACC中研究数据较少。本研究中MYB及NFIB断裂位点与既往检测断裂区域不全相同,不同断裂位点的结合对MYB表达的影响、断裂位点与原发部位的关系仍需进一步探索。MYBL1基因是MYB基因家族中的一员,Mitani等[14]研究发现MYB⁃NFIB融合阴性的唾液腺ACC标本中有35%发生MYBL1⁃NFIB 融合。本研究中21.43%的患者检测到MYBL1⁃NFIB融合,MYBL1断裂区域为exon8,在未检测到MYB 突变的患者中发现MYBL1⁃NFIB 融合的比例较Mitani 研究高,考虑与样本量相关。
在肺MEC患者中,最常见突变为MAML2融合,与既往研究相符。融合的MAML2基因可不通过外源性信号直接激活Notch 信号转录,参与肿瘤细胞的转化、增殖[15]。有学者通过研究胸腺MEC患者的生存率,认为MAML2 融合阳性的MEC 患者较阴性患者中位生存期更长[16],也有文献认为MAML2 基因与预后的相关性不显著,但可协助诊断[17]。本研究随访时间较短,截至2021 年1 月31 日的随访,所有肺MEC患者均存活,因此该基因突变与预后的相关性仍需更多的数据验证。但目前研究发现CRTC1⁃MAML2融合的患者,因EGFR配体双调蛋白的合成上调,使用表皮生长因子受体酪氨酸激酶抑制剂(EGFR⁃TKI)如吉非替尼,可有疗效[18]。
EWSR1 基因突变既往未在MEC 患者中报道,有体外研究发现EWSR1蛋白可以和11 号染色体的FLI1(friend leukemia virus integration 1)易位形成融合蛋白EWS⁃FLI1参与肿瘤细胞的增殖、生长[19],或者与CREM(cyclic AMP element modulator)基因融合协助原发性颅内间叶肿瘤的诊断[20],但与肺MEC的发生发展是否存在相关性尚不明确;且本研究中的PMEC患者于随访时原发灶均未出现复发、转移,该基因可能成为新的生物标记点,但需要更多数据验证。ROS1 基因重排既往在非小细胞肺癌中发生率为1%~2%[21],该基因的检测为靶向治疗提供依据,因ROS1激酶和ALK激酶有49%的氨基酸序列同源性,三磷酸腺苷(adenosine triphosphate,ATP)结合位点有77%同源性[22]。基于Ⅰ期的PROFILE1001 研究,一代ALK抑制剂克唑替尼在2016年已被美国食品及药物管理局(Food and Drug Administration,FDA)和欧洲药物管理局(European Medicines Agen⁃cy,EMA)批准用于治疗ROS1融合的肺癌[23]。
在肺MEC及ACC中均检测到EP300及ARID1A突变。EP300在鳞状细胞癌细胞系研究中被发现与TP53结合后正向调节肿瘤细胞基因表达[24]。研究发现ARID1A 参与肿瘤的发生发展,如在胆管癌中检测到ARID1A 突变通过PI3K/AKT 通路参与细胞转化及肿瘤形成[25],但样本量较小,突变人数较少,上述突变对肿瘤形成的意义仍需更多样本量检测。
在癌组织及癌旁组织差异表达RNA 中,SOX10及BCL11A 的表达上调与ACC 的发生侵袭相关,TFAP2A 对MEC 患者的治疗具有意义。Alex 等[26]在CD133 阳性的ACC 肿瘤干细胞(CSC)中检测到NOTCH1 和SOX10 的高表达,分别敲除NOTCH1 和SOX10,发 现SOX10 和NOTCH1 表达下降。NOTCH1 及SOX10 共同参与ACC 肿瘤细胞的增殖和存活,且相互影响。B 细胞淋巴瘤/白血病11A(B cell lymphom/leukemia 11A,BCL11A)是位于人2 号染色体上的编码锌指蛋白转录因子的基因,调控血红蛋白的成熟,在血液系统疾病中发挥重要作用,后随着研究的深入,发现该基因参与实体肿瘤的增殖侵袭等过程。Lazarus 等[27]通过细胞实验和小鼠模型的结果发现SOX2/BCL11A/SETD8 信号通路可促进气道增生、支气管上皮鳞状化生,导致肺鳞癌形成,其他研究也发现BCL11A 基因通过上游或下游物质参与乳腺癌[28]、前列腺癌等恶性肿瘤的生长、增殖、抗凋亡、转移,在肺涎腺肿瘤中是否存在上述途径仍需体外及体内试验验证。既往研究发现MYB 和BCL11A 均可通过DNA 甲基转移酶1(DNA methyltransferase 1,DNMT1)参与血红蛋白疾病的发生发展[29],但具体的机制尚不明确,是否协同参与肺ACC的发生发展也需进一步的研究,若明确BCL11A 基因的高表达参与肺ACC 的发生发展,该基因可成为肺ACC治疗的重要靶点。
TFAP2A 与血管生成相关,该基因的检测可指导MEC 患者后续抗血管药物的选择。本研究检测到的其他DNA 突变及高表达RNA 因样本量较小,对肺ACC 及MEC 的意义仍需更多的样本量探究。且本研究为回顾性研究,仅存在组织样本,未获取血清或其他组织样本进行对照,后续可结合相关基因结果进行体内试验进一步研究。
猜你喜欢
新民周刊(2022年27期)2022-08-01 07:04:49
今日农业(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
中学生数理化·高一版(2021年2期)2021-03-19 08:32:00
中学生数理化(高中版.高考数学)(2021年1期)2021-03-19 08:28:38
传染病信息(2021年6期)2021-02-12 01:52:58
现代出版(2020年3期)2020-06-20 07:10:34
知识经济·中国直销(2018年8期)2018-08-23 09:16:16
数学学习与研究(2017年3期)2017-03-09 18:12:42
中国老区建设(2016年1期)2016-02-28 09:32:00
