
不同种源蒙古栎遗传变异1)
2022-05-16 04:28李波张伟溪丁密余金金丁昌俊黄秦军
东北林业大学学报 2022年4期
李波 张伟溪 丁密 余金金 丁昌俊 黄秦军
(国家林业和草原局林木培育重点实验室(中国林业科学研究院林业研究所),北京,100091)
蒙古栎(Quercusmongolica)是壳斗科(Fagaceae)栎属树种,极为耐寒,耐旱[1-3]。主要分布于我国东北地区、内蒙古东部、北京、河北等地[4],朝鲜半岛、俄罗斯远东、蒙古及日本北海道等地亦有分布。蒙古栎是我国主要用材树种之一,且具有良好的生态利用价值[5]。蒙古栎研究价值较高,是国家二级保护树种。近年来其空间分布格局、群落结构特征、生长形态特征等研究较多[6-9]。随着生物技术的发展,基于分子标记技术对蒙古栎遗传多样性的研究不断深入,如张杰等[10]应用ISSR标记技术对蒙古栎多种群遗传多样性进行研究,以期为蒙古栎早期选择提供合理依据;有学者通过采用SSR标记技术发现中国东北长白山附近的辽东栎种群是蒙古栎的基因渗入的结果,证明了辽东栎和蒙古栎在该地区是有一定的历史联系[11];有学者[12]利用SNP技术对蒙古栎的遗传多样性进行分析,发现其遗传距离和空间距离之间没有显著相关性。
高通量测序技术的飞速进步,由1~6个核苷酸串联重复的DNA序列的简单序列重复(SSR)得到了广泛的挖掘和开发。不同物种的EST序列和基因组等数据的飞速增加,使开发SSR引物的来源更加丰富。如贯春雨等[13]通过NCBI库下载落叶松属、黄杉属、冷杉属和松属合计共EST序列40 608条,设计EST-SSR引物132对;杨青松和赵艳[14]基于川滇高山栎(Quercusaquifolioides)的转录组数据对其SSR位点分析,结果显示,SSR类型主要为单核苷酸重复;石晓蒙等[15]通过利用栎属62对SSR引物,对栓皮栎(Quercuspetraea)的遗传多样性进行检测,发现其中17对引物具有高多态性;孙静静[16]利用辽东栎(Quercuswutaishansea)和蒙古栎转录组数据开发出92对引物,其中77对能在栎属槲栎组(Quercusaliena)中成功扩增;Ueno et al.[17]以蒙古栎内侧树皮为试验材料,构建了cDNA库,发现274个SSR位点。而通过蒙古栎EST数据库中开发SSR并对群体遗传多样性进行检测的研究相对较少。本研究通过下载蒙古栎EST序列及其叶绿体基因组,对其进行处理后查找SSR位点信息并进行特征分析,设计SSR引物后进行有效筛选,并对不同种源蒙古栎群体进行遗传变异分析,以期为蒙古栎遗传多样性研究提供有效的分子标记技术支持。
1 研究区概况
2018年收集8个不同种源的蒙古栎种子,种植于辽宁省杨树研究所苗圃地,2020年9月进行取样。基本情况见表1。
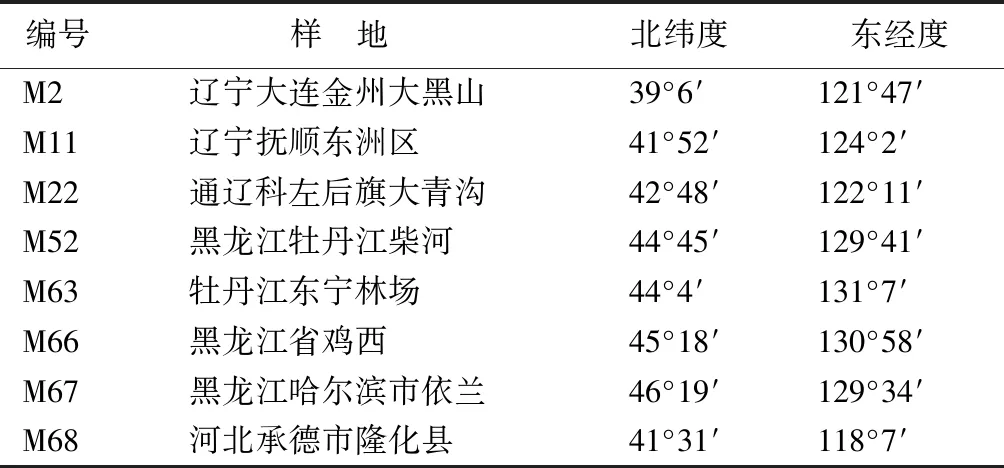
表1 研究样地基本概况
2 研究方法
2.1 DNA的提取
每个种源至少10株,选择其新鲜、完整、无病害的叶片,采用TaKaRa公司DNA提取试剂盒提取蒙古栎基因组DNA,用0.8%的琼脂糖凝胶电泳检测DNA的质量,提取基因组DNA于-20 ℃保存,用于后续PCR扩增。
2.2 EST序列的获得及处理
在NCBI下载蒙古栎EST序列和蒙古栎叶绿体基因组(NC_043858.1)(截至至2019年8月27日)。利用perl脚本est-trimmer,仅用于去除EST序列中过短的序列(<100 bp)以及mRNA的“帽子”和“尾巴”(A或T)。下载CAP3对est-trimmer处理后的序列进行聚类和拼接,以获得无冗余和尽可能长的重叠序列。CAP3的参数取默认值,其中,折叠一致百分比域值N>80,重叠长度域值N>40。
2.3 SSR位点的查找
利用perl脚本misa程序(http://pgrc.ipk-gatersleben.de/misa/misa.html)对CAP3拼接后的序列进行SSR位点搜索。SSR位点重复单元为单核苷酸、双核苷酸、三核苷酸、四核苷酸、五核苷酸及六核苷酸,搜索标准分别对应为,单核苷酸重复数至少为10;双核苷酸重复数至少为6;三核苷酸、四核苷酸、五核苷酸及六核苷酸为重复单位时,重复数均至少为5;以及中间间隔不超过100 bp的复合型SSR。
2.4 SSR引物的设计及筛选
基于Linux(版本bio-linux)Primer3批量设计引物,引物设计的主要参数为:SSR位点上下游100 bp之内;引物长度18~23 bp,20 bp最佳;GC所占比例40%~60%;扩增产物预期片段为100~300 bp,上下游引物解链温度Tm值差异不超过5 ℃。用Electronic PCR去除有多处比较的引物,保证引物扩增的特异性。
PCR反应体系为25 μL:12.5 μL Premix TaqTM(TaKaRa TaqTMVersion 2.0 plus dye)、DNA30~50 ng、正反引物各1.0、9.5 μL双蒸水。PCR反应条件为:预变性95 ℃ 5 s;95 ℃变性30 s,最佳退火温度45 s,72 ℃ 45 s,35个循环;72 ℃延伸10 min,4 ℃保存。PCR产物用2%琼脂糖凝胶电泳进行检测。
2.5 SSR遗传多样性
基于选择的引物进行合成,在样品上进行扩增检测。将所有样品Index PCR扩增产物等量混合,并经割胶回收获得最终的FastTargetTM测序文库,文库的片段长度分布经Agilent 2100 Bioanalyzer验证。文库摩尔浓度精确定量后,最终于Illumina Hiseq平台,以(2×150)bp与(2×250)bp的双端测序模式进行高通量测序,获得Fast Q数据。通过比对读取片段和序列数据计算SSR等位基因的数量。两步校正后列出了SSR基因序列和重复数,包括滑移调整和扩增效率调整。利用软件GenALEX6.501计算等位基因数、期望杂合度、多态性信息含量等指标,采用MEGA version5.0进行群体间UPGMA聚类分析。
3 结果与分析
3.1 蒙古栎EST-SSR分布特点
下载的蒙古栎3 385条EST序列,共1 855 072 bp,平均长度548.03 bp,平均GC含量45.90%。其经est-trimmer和CAP3处理后得到418条无冗余EST序列,全长309 802 bp。共检索出含有SSR位点的序列132条,SSR位点总数为163个;仅含有1个SSR位点的有103条,占78.03%;而含有1个以上SSR位点有29条序列,占21.97%。这些序列中含有18个重叠复合型SSR位点,占总SSR位点的11.04%。
3.2 蒙古栎EST-SSR的类型和频率
蒙古栎EST序列中发现的SSR位点共有5种类型,如表2。其中处理后的EST序列未查找到五核苷酸类型的SSR位点。SSR位点的数量随着核苷酸的数量呈现先增后减的趋势。当重复类型为二核苷酸的时候,SSR位点有80个,达到总SSR位点数的49.08%。而出现频率最低的五核苷酸数量为0,其次就是六核苷酸仅占比0.61%。
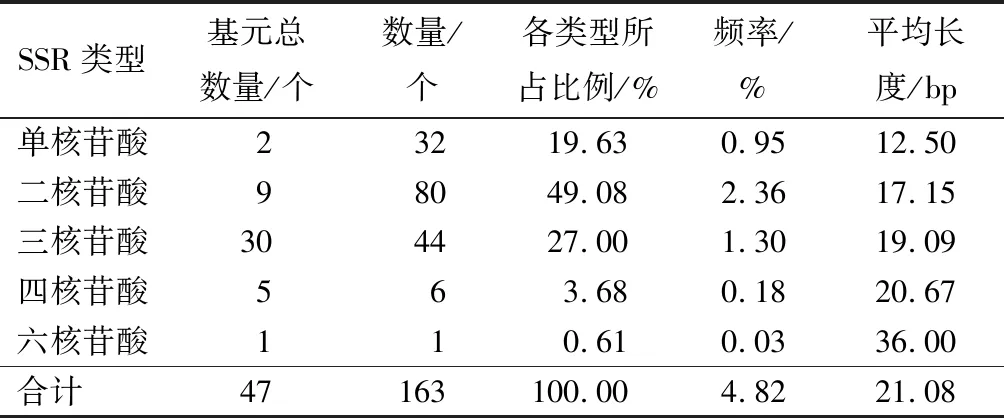
表2 不同SSR重复类型的数量
在蒙古栎的EST序列中,共发现19种SSR重复类型。单核苷酸仅有A与T一种,共有32个,占总SSR重复类型的19.632%。二核苷酸有4种类型,分别是AC与GT、AG与CT、AT与AT、CG与CG,其中AG与CT类型在整个SSR类型中出现频率最多,数量为72个,占比为44.172%。三核苷酸有10种类型,主要是AAC与GTT、AAG与CTT、ACC与GGT、AGG与CCT这4种,而其它6种类型,合计15个,仅占9.201%。四核苷酸仅出现3种类型,AAAC与GTTT、AAAG与CTTT及ATCC与ATGG各出现2次,均占总SSR重复类型的1.227%。六核苷酸AAGCAG与CTGCTT仅出现1次,占总SSR重复类型的0.613%(表3)。
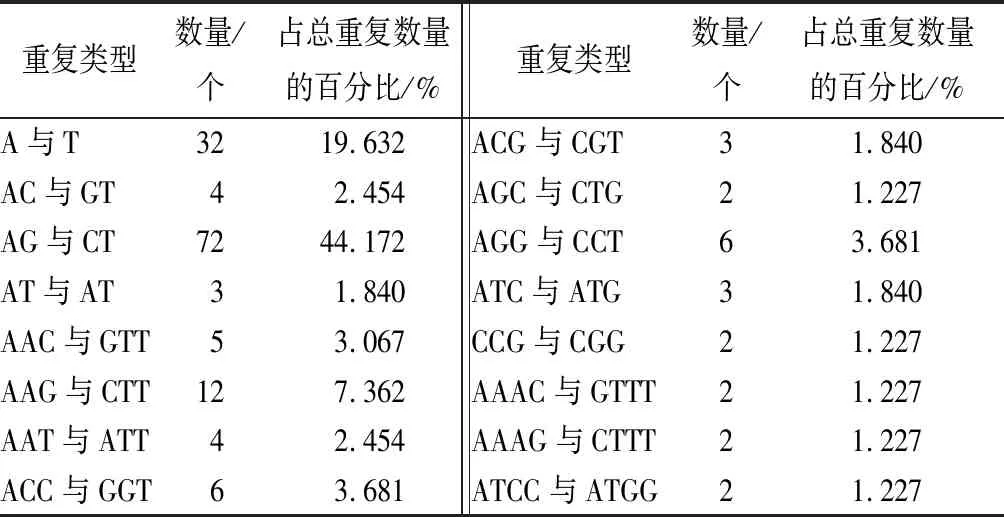
表3 主要SSR重复类型的比例
3.3 叶绿体基因组的SSR特点
下载的蒙古栎叶绿体全基因组共161 194 bp,MISA位点查找SSR位点88个,平均1831.75出现1个SSR位点。88个位点中含有9个重叠复合型SSR位点,占10.230%。叶绿体基因组中SSR类型较为单一,仅有单核苷酸、二核苷酸及三核苷酸。而重复类型为单核苷酸,SSR位点有82个,达到叶绿体基因组总SSR位点数93.18%。二核苷酸重复类型有5个,三核苷酸所占比例极低,仅出现1个,占比1.136%。
从叶绿体基因组SSR的重复次数来看,重复10次和11次数量最多,分别为44个和20个;最多重复次数为15次,共2个。根据MISA查找发现共有4种SSR重复类型。单核苷酸A与T出现频率最高,达81次,占SSR位点总数的92.045%;而单核苷酸C与G仅在重复次数为11次时,出现1个。二核苷酸中出现类型为AT与AT,在重复次数为5、6、7,分别为2个、2个、1个。三核苷酸AAT与ATT出现频率极低,仅在重复6次时,出现1个(图1)。
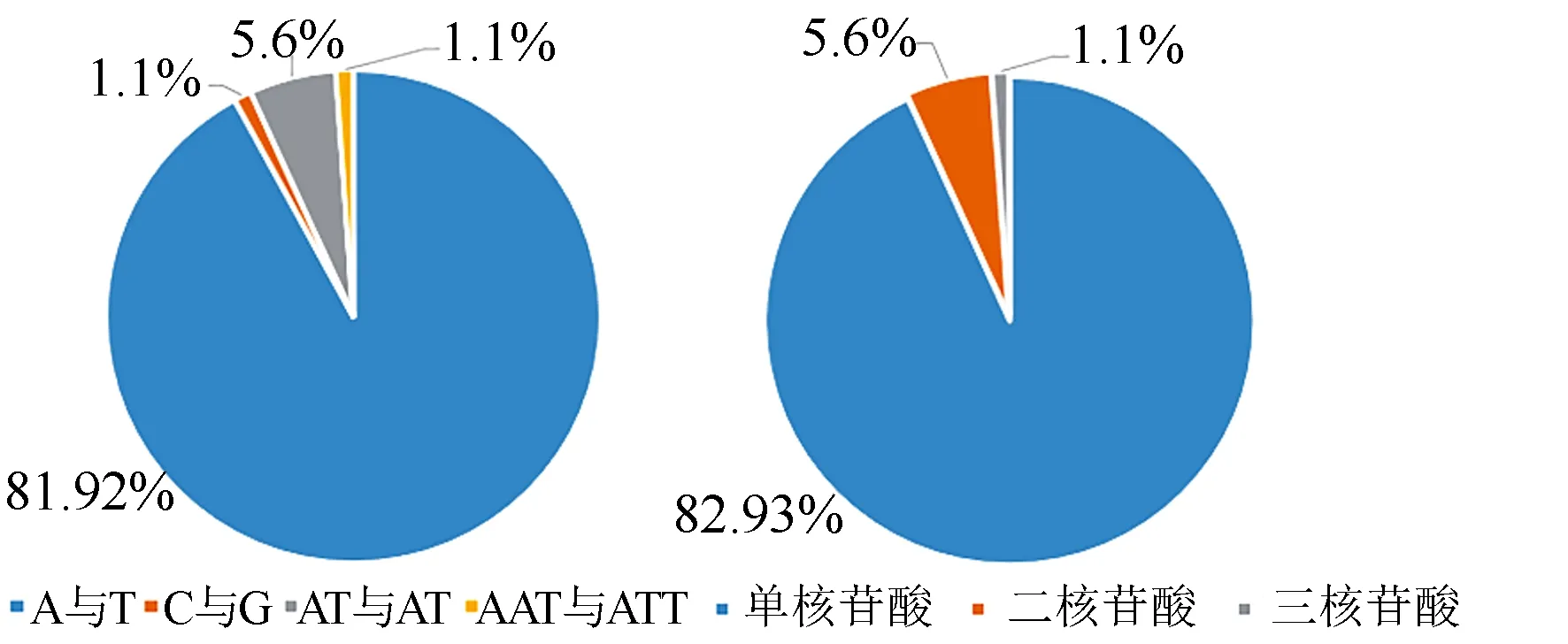
图1 蒙古栎叶绿体SSR的基序类型数量分布
3.4 蒙古栎SSR引物设计及验证
基于perl脚本,整合了MISA(查找SSR位点)、Primer3.0(批量设计引物)和Electronic PCR进行批量SSR引物设筛选计,利用EST序列共设计筛选出引物63对,占总的已鉴定出SSR位点的38.65%;叶绿体全基因组设计筛选30对引物,占总叶绿体鉴定出SSR位点的34.09%(表4)。
对已设计出的引物,进行合成,并在来自不同种源的蒙古栎种源进行PCR扩增验证。PCR扩增结果发现,基于EST序列设计的SSR引物在8份来源不同的DNA上均能扩增出单一条带且扩增效果好,共12对,而扩增出至少6个清晰条带的共有33对(如图2)。基于叶绿体全基因组设计的SSR引物均能扩增出条带且扩增清晰单一,共有13对引物,而能在8个种源DNA中扩增出至少6个清晰条带的有19对。部分引物琼脂糖凝胶电泳图如。
3.5 验证蒙古栎SSR多态性检测
基于上述筛选的结果合成6对引物,从前人对蒙古栎SSR遗传多样性研究中挑选一对引物[18]进行合成。对8个种源群体DNA进行PCR扩增,用以检测这8个种源群体的遗传多样性以及引物多态性,引物信息见表4。哈温平衡检测发现7个位点在8个种源蒙古栎群体上均未偏离平衡(表5)。7对SSR引物在8个种源的蒙古栎群体中共检测到37对等位基因,这些位点的观测等位基因数目为2.5(SH-21)~7.625(Qden05011),平均每个位点有5.286个等位基因(Na)。有效等位基因(Ne)的变化范围为1.637(SA-23)~5.118(SA-43),平均有效等位基因数为3.643。从Shannon多样性指数看,SH-21的值最小(I=0.64),Qden05011的值最大(I=1.801),平均Shannon多样性指数为1.291。观测杂合度(Ho)和期望杂合度(He)的变化范围分别为0.315(SH-21)~0.847(SA-39)、0.381(SA-23)~0.799(Qden05011),均值分别为0.605和0.622。多态性信息含量的变化范围为0.371(SA-23)~0.893(SA-49),均值为0.692,从多态性看,SA-39、SA-43、SA-49与Qden05011的多态性水平相差不大。

表4 多态性SSR引物信息

引物SA47、SA48、SA49分别对应1~8、9~16、17~24。

引物SC24、SC25、SC26分别对应1~8、9~16、17~24。
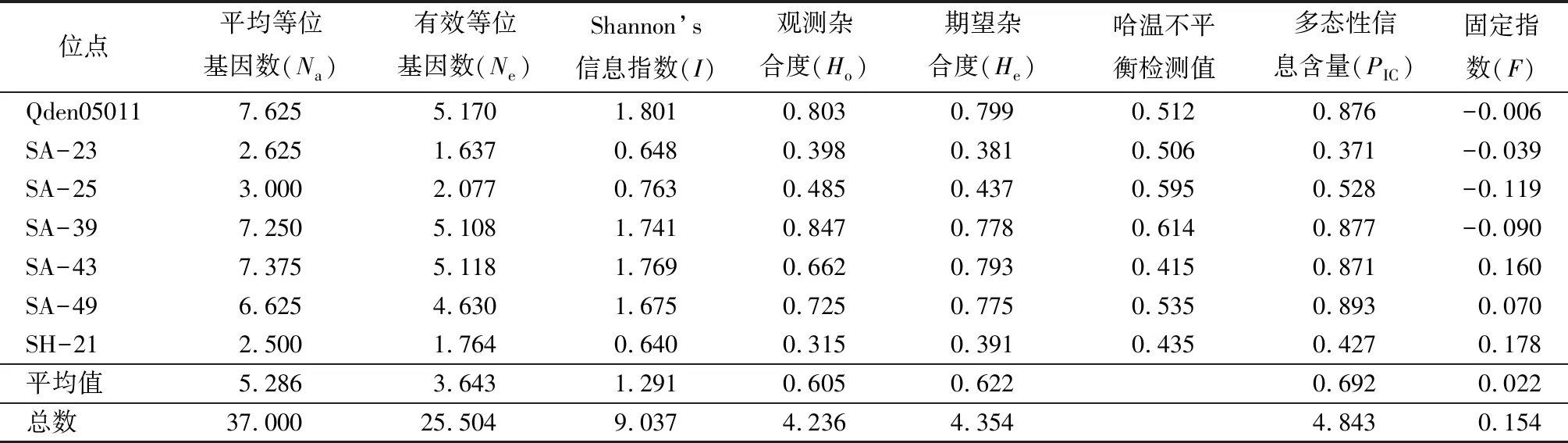
表5 7个SSR位点在蒙古栎群体中的遗传多样性参数
8个种源蒙古栎群体水平的遗传多样性见表6。不同种源群体的观测等位基因(Na)和有效等位基因数(Ne)的变化范围分别为4.571(M2和M66)~6(M63)、2.976(M67)~4.373(M63),平均值分别为5.286和3.643。Shannon多样性指数的均值为1.291,其中M2的值最小(I=1.107),M68的值最大(I=1.495)。观测杂合度(Ho)和期望杂合度(He)的变化范围为0.546(M63)~0.698(M68)、0.538(M2)~0.71(M68),无偏期望杂合度(UHe)的均值为0.66,其变化范围为0.57(M2)~0.752(M68)。M66的固定指数(F)最大,为0.167,且其期望杂合度高于观测杂合度,说明种源M66存在遗传缺失现象。M11的固定指数最小,仅为-0.101,而它的期望杂合度低于观测杂合度,说明该种源内部个体间存在遗传分化的现象。
根据等位基因数据计算其群体内近交系数(Fis)、群体间近交系数(Fit)、群体间遗传分化系数(Fst)和基因流(Nm),见表7。群体内近交系数的均值为0.025,SA-25位点的值最小,为-0.11,SH-21位点的值最大,为0.192;群体间近交系数的均值为0.168,SH-21位点的值最大,为0.382,而最小值为0.015(SA-23);群体间遗传分化系数的均值为0.147,变化范围为0.056(SA-23)~0.279(SA-25),说明蒙古栎群体间具有中等水平的遗传分化。基因流的变化范围为0.647(SA-25)~2.264(Qden05011),均值为1.933,说明蒙古栎群体间存在较大的基因流,减弱了基因遗传漂变的作用,群体间发生遗传分化的程度有所减弱。
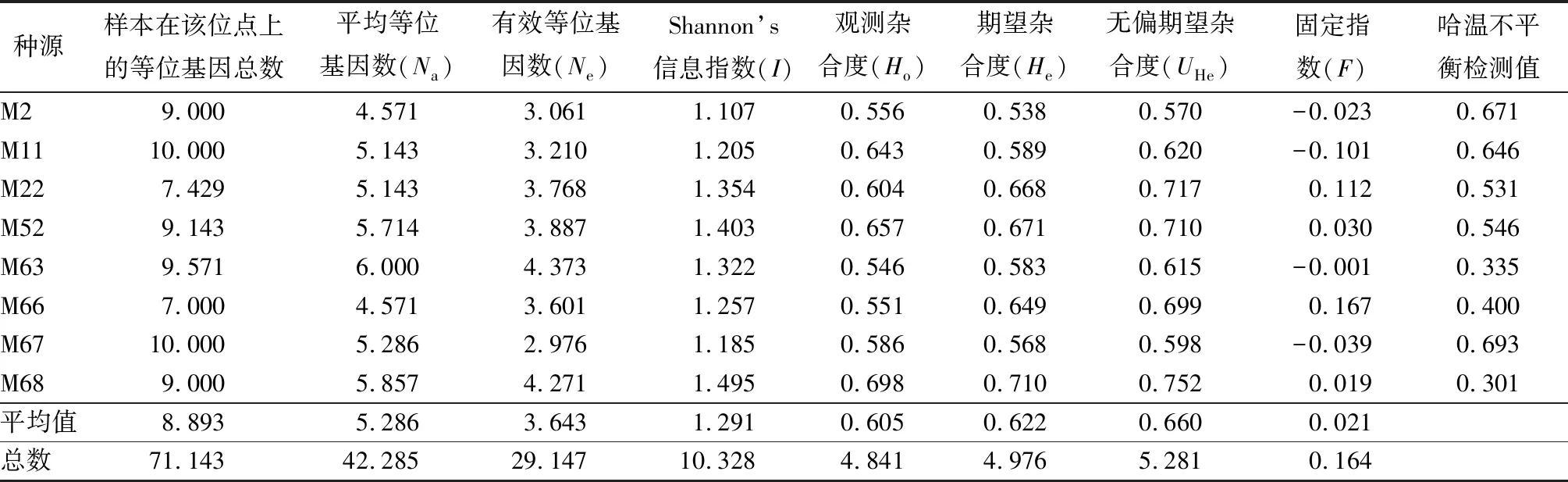
表6 蒙古栎群体水平的遗传多样性参数
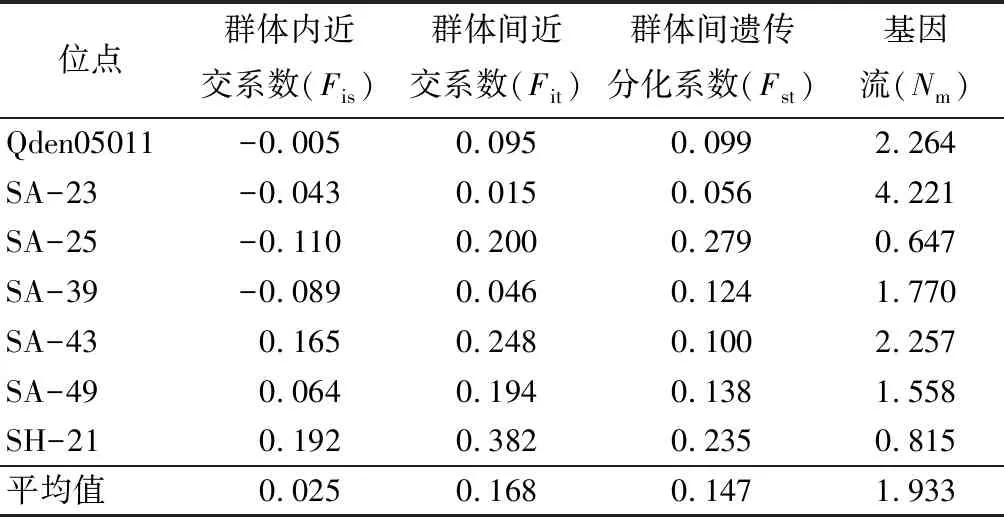
表7 蒙古栎群体遗传分化系数和基因流
对蒙古栎群体的AMOVA分析结果(表8)表明:种源间的遗传变异占10.216%,种群内的变异占89.784%,与种源群体间遗传分化系数相差不大,说明了蒙古栎的遗传多样性主要是由种源群体内的遗传差异导致的。根据Nei’s遗传距离构建蒙古栎不同种源的UPGMA聚类图,见图3。
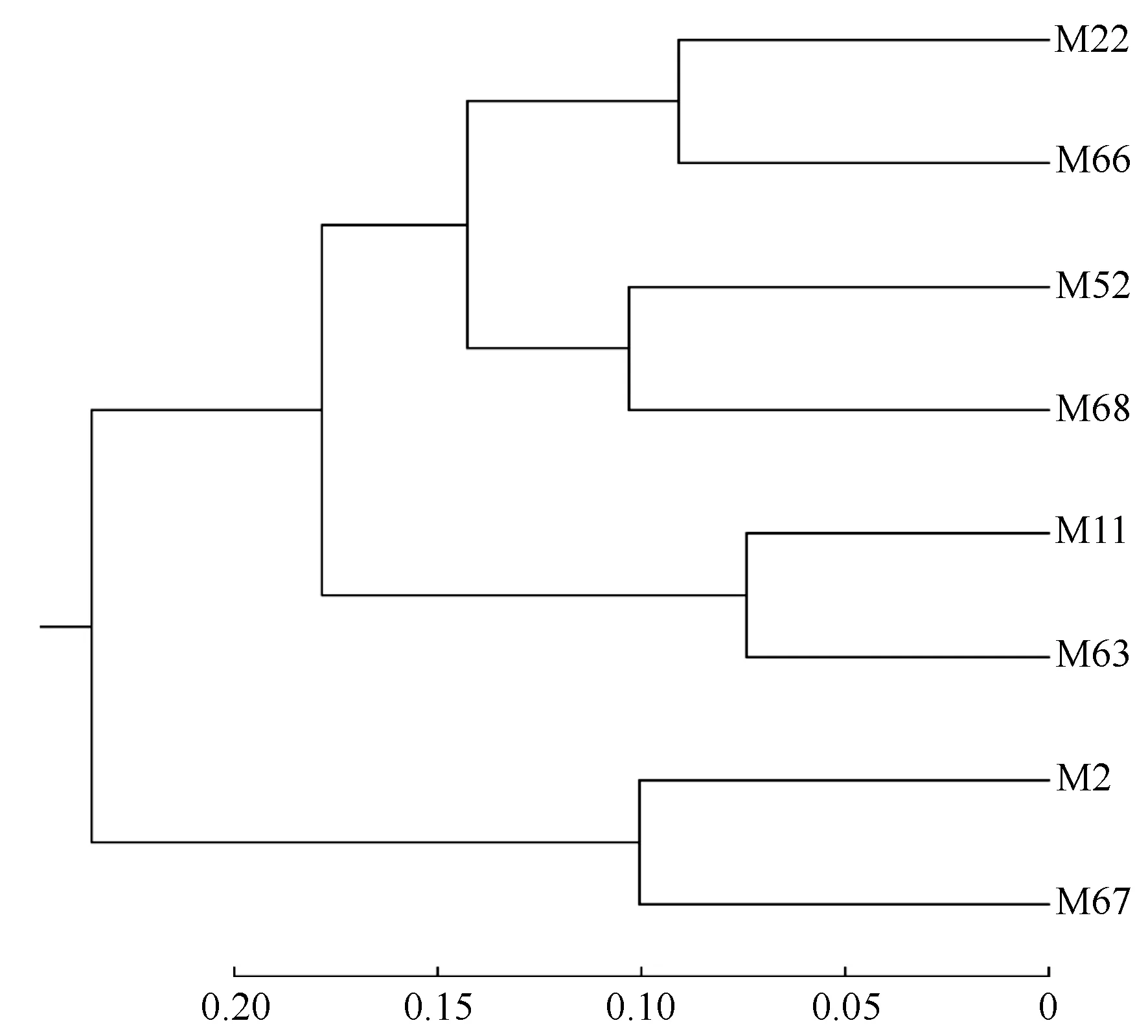
图3 蒙古栎群体UPGMA遗传距离聚类图
8个种源的蒙古栎可分为3类,第1类:M22、M52、M66和M68;第2类:M11和M63;第3类:M2和M67。可以看出,蒙古栎种源与地理位置可能存在较弱的相关性,对遗传距离和地理距离进行Mantel检验,发现两者没有显著相关性(R2=0.001 8,p=0.09),结果表明:地理隔离不是导致蒙古栎遗传变异的主要因素。
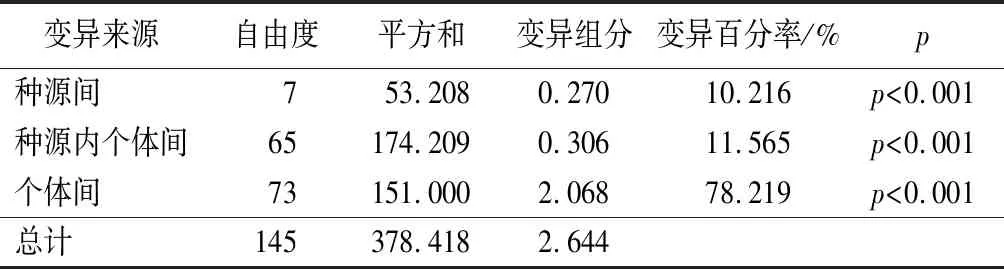
表8 蒙古栎种源群体的分子方差分析
4 结论与讨论
随着测序技术的快速发展,SSR分子标记的引物开发方法也随之变多。费时费力构建传统文库逐渐被新的技术方法取代,如通过改良文库法,即构建富集文库测序开发SSR引物[19];基于该物种转录组数据或者通过简化基因组测序进行开发SSR引物的研究越来越多[20-21]。而随着数据库EST和全基因组的增加,开发SSR引物也越来越方便,即通过下载数据库中的EST序列,处理后通过软件或脚本查找SSR位点信息,再使用引物设计软件进行设计筛选引物[22-24]。
而SSR位点查找软件或脚本,大多研究使用perl脚本MISA进行查找[24-26]。还有通过李强等[27]开发的本地SSR Hunter进行本地SSR位点搜索查找。但二者一般搜索标准不同,MISA一般设置条件为:单核苷酸为重复单位时,重复数至少为10;双核苷酸为重复单位时,重复数至少为6;三核苷酸、四核苷酸、五核苷酸及六核苷酸为重复单位时,重复数均至少为5。而本地软件SSR Hunter的一般标准为:重复单位长度为2~6 bp,最少重复次数为5次。搜索标准因人而异,但是SSRHunter的不同之处在于,对单核苷酸重复位点不能进行查找,不能对中间间隔不超过100 bp的复合型SSR进行筛选,但是这款软件查找位点信息可以给出位点上下游150 bp信息,便于逐一设计SSR引物。而MISA可通过perl脚本结合Primer3进行批量设计引物,显然更快捷方便。
本研究通过SSR Hunter 1.3查找SSR位点175个,逐一设计筛选比对得到引物22对,在8份种源不同的模版上有效扩增达6份以上,且单一性良好、条带清晰的有14对引物,达63.64%;而MISA查找位点163个,批量设计筛选引物63对,8份模版上有效扩增达6份以上,且单一性良好条带清晰的有33对,占比52.38%。因为数据工作量的重复性和批量设计引物的需求,相比较MISA更方便。对比本研究结果,张元燕等[28]对4种栎属的SSR位点进行分析,序列处理采用的软件及SSR位点查找标准的不一致,会导致有明显差异。
通过从NCBI数据库中下载蒙古栎EST序列,经过处理组装得到418条序列,全长309 802 bp,含有SSR位点有132条序列,SSR位点163个。而蒙古栎叶绿体基因组全长161 194 bp,SSR位点88个。相比较其他物种,数据库中蒙古栎EST序列较少,可能与蒙古栎研究较少有关。基于蒙古栎EST序列发现的SSR类型,单核苷酸、二核苷酸和三核苷酸较多,达到95%以上;而五核苷酸则没有发现,六核苷酸重复类型仅占比0.61%。基于叶绿体基因组查找的SSR,仅存在单核苷酸,二核苷酸和三核苷酸,单核苷酸最多,88个SSR位点中出现82个单核苷酸位点。19种重复类型中,AG与CT和A与T,这二种类型占了总SSR重复类型的63%。二核苷酸AG与CT出现的比例最高,出现72次。这与王书珍等[29]的研究结果相似,二核苷酸最多,其次是单核苷酸。徐小彪等[30]研究发现猕猴桃EST-SSR中二核苷酸为主要类型,AG与CT出现次数最多,结果相似。在叶绿体基因组中,A与T出现比例最高,为81次。重复次数为10次和11次时,SSR位点出现最多。
设计合成的6对SSR引物,与前人研究的Qden05011引物相比较,在这8个种源群体上,SA-39、SA-43、SA-49与Qden05011的多态性水平一致,具有较高的多态性。有学者[31]利用9对SSR引物对蒙古栎的多态性进行检测,结果显示其多态性信息含量的变化范围为0.487 2~0.848 3,均值为0.723 7,孙静静[16]设计的SSR引物PIC的变化范围为0.49~0.95,均值为0.83。本研究开发的6对SSR引物的多态性信息含量为0.371~0.893,均值大于0.5,说明了开发的SSR引物在蒙古栎遗传多样性研究中起到的作用是显著的。
7对引物在8个不同种源蒙古栎上的遗传多样性分析结果显示,每个SSR位点的平均等位基因数(Na)为3.643,Shannon多样性指数(I)为1.291,平均期望杂合度(He)为0.622,平均多态性信息含量(PIC)为0.692。张杰[32]利用ISSR技术分析蒙古栎的遗传多样性,结果为Na=1.451 8、Ne=1.305 9和I=0.254 6;陈罡等[33]利用10对SSR分子标记对分布在辽宁省的蒙古栎天然群体进行遗传多样性分析,结果显示,Na=7.3、I=1.270、He=0.624,这说明了蒙古栎群体具有较高的遗传变异和遗传多样性水平。对比栎属其他树种,辽东栎(Na=10.272 7、I=1.754 3、He=0.753 8)[34]、白栎(Quercusfabri)(Na=5.41、He=0.542、I=1.151)[35]、短柄枹栎(Quercusglandulifera)(Na=3.67、He=0.43、I=0.66)[36]、欧洲栓皮栎(Quercusvariabilis)(He=0.65、Na=0.72)[37],说明蒙古栎存在较为丰富的遗传多样性,在栎属树种中具有中等程度的遗传多样性。
种源间的遗传多样性分析结果显示:不同种源间存在一定的差异性,种源间存在遗传缺失或种源内部间个体间遗传分化等现象。李文英等[38]采用AFLP技术分析蒙古栎群体遗传变异,发现蒙古栎天然群体间存在一定程度的遗传分化(Gst=0.077),且存在一定的基因交流,Nm为5.99;李文英和顾万春[39]利用等位酶分析蒙古栎天然群体发现其群体间遗传分化度Gst为0.107,群体内变异量占89.27%,但其基因流Nm为2.08,处于较低水平;张杰等[10]利用ISSR分析蒙古栎群体遗传多样性发现,种源内的遗传变异占总遗传的73.43%,基因流Nm值较低,仅1.381 8;王越[18]利用SSR标记分析蒙古栎遗传多样性,结果显示94.41%的遗传多样性存在于种群内,基因流为4.222 1,蒙古栎群组的遗传分化程度处于中等水平;陈罡等[33]开发10对SSR引物对辽宁蒙古栎8个天然群体分析发现,群体间遗传分化系数为0.065 5,遗传变异主要存在于群体内,群体间存在的基因流达4.471。本研究发现蒙古栎群体的基因流为1.933,处于较低的水平。其基因流较低的原因可能是风媒传粉的多年生蒙古栎[40],其种子主要依靠重力和小型啮齿动物传播,因而较大的地理隔离可能会导致种源群体间的低基因流;生境片段化亦会影响基因流的强度。弱基因流仅在一定程度上弱化种间的遗传差异,减少群体间的遗传差异,因而本研究发现F统计显示群体间的遗传分化系数仅为0.147,相对较高,且AMOVA分析也发现群体间的遗传变异占10.216%,种源内的变异占89.784%。相较其他栎属的研究,舒玛栎(Quercusshumardii)群体间变异贡献率达到15.12%[41]、意大利卡拉布里亚的栓皮栎13%的变异存在于群体间[9]、意大利南部栓皮栎20%的遗传变异存在于居群间[9]、大果栎(Quercusmacrocarpa)3%的遗传变异发生在居群间[9]、川滇高山栎居群间的变异为8.9%[42]、槲栎11.45%的变异是发生在群体间的[16],蒙古栎种源间分化水平处于中等水平程度。利用Mantel检验发现蒙古栎遗传距离和地理距离之间没有显著相关性,且根据聚类分析发现蒙古栎群体并非按照地理位置聚类,这与李文英和顾万春[39]的结果一致,同样的结果也出现在其他栎属树木中,栓皮栎[43]、麻栎(Quercusacutissima)[44]、白栎[45]等,这可能跟其自身遗传特性相关,且生存环境变化有关。
采用不同SSR位点查找软件,因其不同的搜索标准,可能产生的结果有所不同。在大量转录组或者数据库数据下,快速便捷的MISA结合Primer3、Electronic PCR批量设计并筛选去除有多处比较的引物,明显更适合研究的需求。基于蒙古栎已知EST数据库进行处理查找SSR位点,发现二核苷酸和单核苷酸发生频率最高,而叶绿体基因组中单核苷酸发生频率最高。设计合成的93对引物,其中有52对引物能在多种源中有效扩增,为蒙古栎遗传多样性研究和分子标记辅助育种奠定了基础,提供了构建遗传谱系的依据;基于7对SSR引物发现,蒙古栎具有中等程度的分化水平,且主要的遗传变异来源群体间,个体间存在遗传分化或者缺失现象。蒙古栎群体并非完全按照地理位置聚类,可能与其自身遗传特性和生存环境变化有关。
猜你喜欢
农村科学实验(2022年15期)2022-10-13
肝博士(2022年3期)2022-06-30
承德医学院学报(2022年2期)2022-05-23
福建林业科技(2022年1期)2022-04-28
中国典型病例大全(2022年9期)2022-04-19
昆明医科大学学报(2022年2期)2022-03-29
中国体育教练员(2021年4期)2022-01-21
烹调知识(2019年3期)2019-03-01
吉林农业(2016年7期)2016-05-14
江苏农业科学(2014年6期)2014-08-12
