
中药渣堆肥微生物群落结构及纤维素降解酶基因表达量变化特征
2022-05-16 17:34杜婕宋修超马艳
江苏农业学报 2022年2期
关键词:群落结构
杜婕 宋修超 马艳
摘要:本研究對不同堆制时期的中药渣堆肥样品进行Miseq高通量测序分析,并对8种纤维素降解酶基因进行荧光定量RT-PCR分析,以解析中药渣堆肥过程中微生物群落结构组成及纤维素降解酶基因表达丰度的变化特征。结果表明,在中药渣堆肥过程中,不同堆制时期的微生物群落结构组成不同,并且不同处理间的群落结构也存在差异。对样品中微生物群落进行主成分分析(PCoA),发现在中药渣堆肥过程中,不同处理的组间微生物群落结构存在较大差异,组内群落结构不断变化并逐渐趋于稳定。荧光定量RT-PCR检测结果显示,在中药渣堆肥过程中,纤维素降解酶基因的表达丰度也处于明显的变化中。说明,中药渣堆肥中微生物群落与纤维素降解酶是相互联系、相互影响的。
关键词:中药渣堆肥;Miseq高通量测序;群落结构;纤维素降解酶基因
中图分类号:S141.4文献标识码:A文章编号:1000-4440(2022)02-0352-09
Change characteristics of microbial community structure and cellulose degrading enzyme gene expression level in Chinese medicine residue compost
DU Jie SONG Xiu-chao MA Yan
(1.Institute of Agricultural Resources and Environment, Jiangsu Academy of Agricultural Sciences, Nanjing 210014, China;2.Key Laboratory of Agro-Environment in Downstream of Yangtze Plain, Ministry of Agriculture and Rural Affairs, Nanjing 210014, China;3.School of the Environment and Safety Engineering, Jiangsu University, Zhenjiang 212013, China;4.Institute of Animal Husbandry and Veterinary Medicine, Jiangsu Vocational College of Agriculture and Forestry, Zhenjiang 212400, China)
Abstract: In this study, Miseq high-throughput sequencing analysis was performed on samples of Chinese medicine residue compost during different composting periods, and eight kinds of cellulose degrading enzyme genes were analyzed by fluorescence quantitative RT-PCR to explore the composition of microbial community structure and the expression and abundance of cellulose degrading enzyme genes during the composting of Chinese medicine residues. Results showed that during the composting of Chinese medicine residues, the composition of microbial community structure in different periods was different, and the community structure was also different in different treatments. Principal component analysis (PCoA) results indicated that during the composting of Chinese medicine residues, there were significant differences in the microbial community structure among different treatments, and the community structure within the group changed continuously and gradually stabilized. Results of fluorescence quantitative RT-PCR showed that the expression abundance of cellulose degrading enzyme genes also changed significantly during the composting of Chinese medicine residues. In conclusion, the microbial community and cellulose degrading enzymes in Chinese medicine residue compost are interrelated and interacted.
Key words:Chinese medicine residue compost;Miseq high-throughput sequencing;community structure;cellulose degrading enzyme genes
中药渣是中药材在加工、炮制过程中以及中成药生产过程中,或者其他中药材相关产品(如保健产品)加工过程中产生的废弃物[1-2]。近年来,随着中医药卫生事业的发展以及人们健康意识的增强,中药的生产、开发力度逐年增长,导致中药材生产、加工后产生的药渣也越来越多。中药渣里含有丰富的木质素、纤维素、半纤维素,大量的多糖、蛋白质、氨基酸等生物活性物质以及多种微量元素[3-4],具有重要的再利用价值。目前大多采取焚烧、堆放、填埋等简单粗放的方式对中药渣进行处理,不仅占用土地、污染环境,还会造成严重的资源浪费。堆肥化处理技术是国内外广泛采用的一种有机固体废弃物资源化利用技术。对中药渣进行堆肥化处理是既能减少污染,又能对其进行资源化利用的处理方式。中药渣堆肥中因含有大量的木质素、纤维素等而具有较长的腐熟周期,因此利用高效、环保的微生物法对木质素、纤维素进行分解和生物转化是实现中药渣资源化利用的有效途径。在此过程中,具有纤维素或木质素降解功能的微生物是微生物法实施的关键[5],它们可以将纤维素、木质素等转化为小分子物质,释放药渣中残留的活性物质,有利于实现中药渣中大分子物质的降解[6]。
近年来,宏基因组学技术避开传统微生物的分离培养方法,直接从样品中提取总DNA进行分析,可以更完整、准确地反映微生物的群落组成特征。目前,DNA高通量测序已成为现代生命科学领域常用的研究技术[7-10],且近几年迅速发展的DNA高通量测序技术能检测到许多低丰度内生细菌[11],已成功应用于多个领域,如发酵食品、海洋环境、肠道环境、植物多样性、人工湿地微生物[12-13]。高通量测序技术在奶牛瘤胃菌群结构多样性[14],纤维素降解复合菌系中微生物的群落结构[15]以及关键降解功能菌[16]的研究中有所应用,但利用Miseq高通量测序技术对中药渣堆肥中微生物群落结构组成和变化进行研究的报道较少。本研究拟以中药渣(青蒿药渣和金银花药渣)堆肥为研究对象,分析堆肥过程中微生物群落组成及优势菌群的变化情况,同时采用荧光定量RT-PCR技术检测在此过程中与纤维素降解相关酶基因表达丰度的变化特征,探索中药渣堆肥过程中微生物群落结构和功能基因的变化规律,以期为中药渣等堆肥的降解及资源化利用奠定基础。
1材料与方法
1.1试验材料
中药渣堆肥样品:中药渣(青蒿药渣和金银花药渣)堆制于江苏省连云港东海县。本研究共设3个处理,对照(CK):青蒿药渣∶金银花药渣∶混合一∶混合二=2.00∶1.00∶1.00∶1.00(质量比);T1处理:青蒿药渣∶金银花药渣∶混合一∶混合二∶菌剂=2.00∶1.00∶1.00∶1.00∶0.04(质量比);T2处理:青蒿药渣∶金银花药渣∶混合一∶混合二∶回料=2.00∶1.00∶1.00∶1.00∶0.10(质量比)。在中药渣堆制期间,各处理分别取样10次,每次取3个样品。
本研究所用的主要试剂和仪器:rTaq DNA酶(TaKaRa公司产品)、T4 DNA聚合酶(TaKaRa公司产品)、土壤DNA提取试剂盒(MP Biomedicals公司产品)、胶回收试剂盒(Axygene公司产品)、凝胶电泳仪(BioRad公司产品)、凝胶成像仪(BioRad公司产品)、PCR仪(Eppendorf公司产品)、核酸测定仪(Thermo Fisher Scientific公司产品)、摇床(南京百思禾生物科技有限公司产品)、振荡培养箱(南京百思禾生物科技有限公司产品)、ABI 7500型定量PCR仪(ABI公司产品)。
1.2试验方法
1.2.1中药渣堆肥样品DNA提取利用土壤DNA提取试剂盒(MP Biomedicals公司产品)对3个处理中不同堆制时间的中药渣样品进行DNA提取,通过NanoDrop 2000超微量核酸测定仪(Thermo Fisher Scientific公司产品)对提取的DNA样品进行质量检测。
1.2.2Miseq高通量测序采用Caporaso等[17]提出的方法对中药渣样品中的微生物进行Miseq高通量测序。利用特异性引物515F(5′-GTGCCAGCMGCCGCGG-3′)和907R(5′-CCGTCAATTCMTTTRAGTTT-3′)对样品中细菌16S rRNA基因V4区进行扩增。扩增体系为:Master Mix (2×) 15.0 μl,F/R引物2.0 μl,模板DNA为10.0 ng,然后再用超纯水补齐30.0 μl。PCR程序参照文献[18]进行,采用QIAquick-PCR純化试剂盒对PCR扩增后的产物进行纯化,利用Invitrogen公司的Qubit12.0荧光光度计对纯化产物进行定量分析,纯化后的DNA产物送至上海美吉生物医药科技有限公司利用Illumina-MiSeq仪器[宜曼达贸易(上海)有限公司产品]进行测序。
1.2.3中药渣中微生物信息分析通过拼接、过滤和去除嵌合体3个步骤将Miseq高通量测序得到的序列拼接成优化序列,并进行质量检验。去除标记序列和引物序列,将剩余序列聚合成具有97%相似性序列的操作分类单元(OTUs)。用QIIME1.7.0软件在Greengene数据库(http://greengenes.secondgenome.com/)中对这些OTUs进行注释[19-20]。通过多样性分析,研究不同堆肥时期中药渣中微生物群落结构的差异,同时比较不同处理间中药渣堆肥样品微生物群落的组成结构。
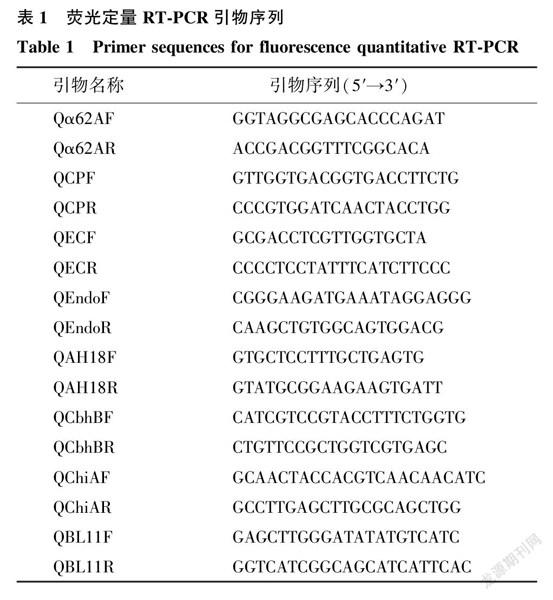
1.2.4堆肥过程中纤维素降解酶基因表达丰度测定根据中药渣堆肥中微生物的种类信息,从NCBI数据库中搜索8种具有纤维素降解功能的降解酶,包括:阿拉伯糖苷酶(α62A)、纤维二糖磷酸化酶(CP)、纤维素水解酶(EC)、内切葡聚糖酶(Endo)、纤维素酶(AH18)、纤维素分解酶(CbhB)、几丁质内切酶(ChiA)、多糖基水解酶(BLII),根据降解酶的基因序列设计特异性引物(表1)进行荧光定量RT-PCR测定,检测堆肥中微生物纤维素降解酶基因表达丰度的变化特征。
通过特异性引物对上述8种纤维素降解酶基因进行扩增,扩增成功的目的片段与载体pEASY-T1连接,然后将其转化入DH5α感受态细胞中。利用通用引物M13F(5′-TGTAAAACGACGGCCAGT-3′)和M13R(5′-CAGGAAACAGCTATGACC-3′)对细菌群落进行扩增,选择阳性转化子送至上海美吉生物医药科技有限公司进行测序验证,序列正确的质粒用于制作标准曲线。
用荧光定量PCR扩增酶对中药渣堆肥DNA样品进行扩增,扩增总体积为20.0 μl,具体为:荧光定量PCR扩增酶10.0 μl,Temp DNA 1.0 μl,F/R引物各1.0 μl,超纯水(ddH2O)为7.0 μl。加样混匀后,将PCR反应溶液置于ABI 7500型定量PCR仪(ABI公司产品)上进行扩增,具体程序为:95 ℃ 5 min预变性;95 ℃ 30 s,55 ℃ 30 s,72 ℃ 45 s,40个循环,72 ℃ 10 min,4 ℃下保存。根据熔解曲线,计算纤维素降解酶基因表达丰度。
1.3数据分析
用SPSS 20.0软件对试验数据进行统计分析。试验数据采用单因素方差分析,用平均值±标准差表示。
2结果与分析
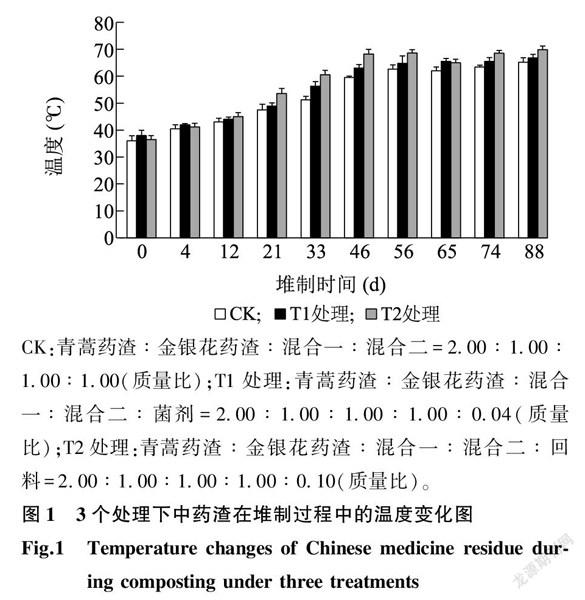
2.1中药渣堆肥腐熟过程中温度变化趋势
图1显示,在中药渣堆制期间,本研究3个处理在堆制46 d时都已经进入高温期(温度≥55 ℃),T2处理的温度最高,甚至可达70 ℃。中药渣堆制88 d时,3个处理仍处于高温期。
2.2不同分类水平上中药渣堆肥中微生物群落组成变化
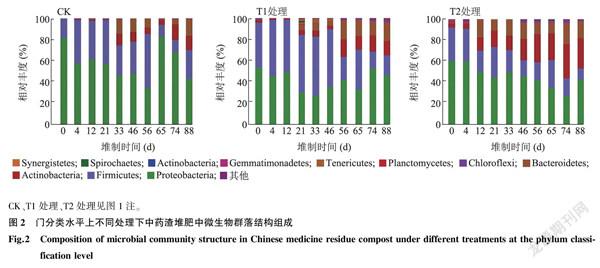
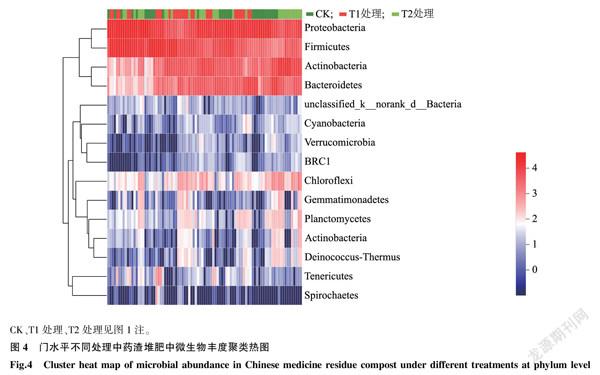
对不同时期的中药渣堆肥样品进行Miseq高通量测序,图2显示,在门水平上,厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)和放线菌门(Actinobacteria)在对照(CK)、T1处理、T2处理中均为优势菌群并且在整个堆制期间一直存在,但随着堆制时间的延长其所占比例有所变化。总体而言,与CK相比,T1处理中厚壁菌门所占比例有所增加;与CK、T1处理相比,T2处理中厚壁菌门所占比例在大多数堆制时间下较小,且随着堆制时间的延长其所占比例总体呈减少趋势。
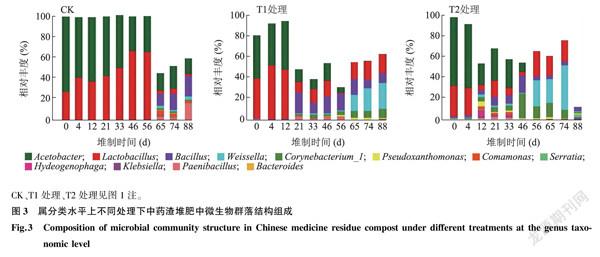
分析微生物在属水平上的群落结构变化,结果(图3)表明,醋酸杆菌属(Acetobacter)在3个处理中占比均较高,为优势菌群,但随着堆制时间延长其所占比例总体呈减少趋势。CK中乳酸杆菌属(Lactobacillus)的占比在堆制0~56 d时随着堆制时间延长总体呈增加趋势,在65~88 d时其所占比例大大减少;在T1处理中,乳酸杆菌属(Lactobacillus)在堆肥0~12 d时占比较大,21~88 d时占比明显减少;在T2处理中,随着堆制时间的延长(0~46 d),乳酸杆菌属(Lactobacillus)总体呈减少的趋势,而在56~74 d时其占比又有所增加。此外,假黄单胞菌属(Pseudoxanthomonas)存在于T1、T2处理中,与CK相比,魏瑟拉属(Weissella)在T1、T2处理中占比较大,且在T1处理中随着堆制时间延长其占比逐渐增加。
2.3中药渣微生物组成分析和主成分分析
微生物群落组成的热图是以颜色梯度代表數据矩阵中数值的大小,并对类型、数量相似的微生物群落进行聚类分析[21]。微生物群落丰度不同,则颜色不同,根据此特征可将样品中的微生物群落进行分类,从而显示群落结构的相似之处和不同之处。在门水平上对中药渣样品中微生物物种组成和丰度进行物种热图聚类分析,同一行中颜色深浅代表物种在样品中的丰度高低,左边聚类表示物种间的相似度。图4显示,3个处理间中药渣的微生物群落组成差异较大。同时,各处理中微生物群落结构组成是不断变化的,且随着堆制时间延长,各处理中微生物群落的丰度差异逐渐变大,微生物优势菌群也发生明显变化。
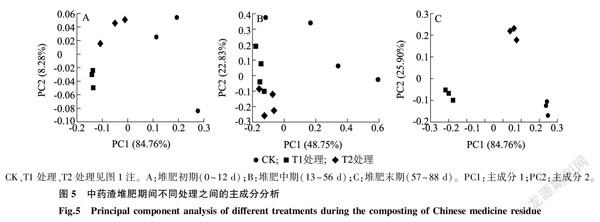
对中药渣堆肥3个处理中微生物群落进行主成分分析(PCoA),结果(图5)表明,随着中药渣堆制时间的延长,CK、T1处理、T2处理中微生物群落特征发生明显变化。堆肥初期(0~12 d,图5A)和堆肥中期(13~56 d,图5B)CK、T1处理、T2处理的微生物群落组成不同,T1处理和T2处理的样品中微生物群落较为集中,因此这2个处理的微生物群落结构较为稳定,但T1、T2处理在堆肥不同时期的群落结构存在差异,处于变化中。在堆肥末期(57~88 d,图5C),CK、T1处理、T2处理的微生物群落结构表现为组内聚在一起,组间分散较大,说明不同处理间群落结构差异更加明显,并且此时3个处理的组内群落结构更为稳定。此外,在同一处理中,微生物群落结构随着堆制时间的延长而发生改变,并且这种改变使组内群落结构逐渐趋于稳定。同时,通过对比堆肥早期、中期、晚期的群落结构,发现在堆肥进程中,3个处理间的微生物群落结构一直存在较大差异,且随着堆制时间的延长,群落结构处于动态变化中。
2.4中药渣堆肥中纤维素降解酶基因表达量变化特征
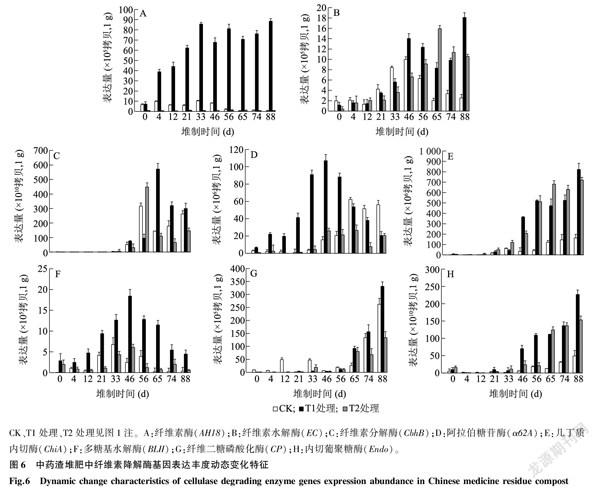
本研究检测了8种纤维素降解酶[阿拉伯糖苷酶(α62A)、纤维二糖磷酸化酶(CP)、纤维素水解酶(EC)、内切葡聚糖酶(Endo)、纤维素酶(AH18)、纤维素分解酶(CbhB)、几丁质内切酶(ChiA)、多糖基水解酶(BLII)]基因在中药渣堆制期间表达量的变化,结果(图6)表明,T1处理AH18基因表达量明显高于CK和T2处理,并随着堆制时间的延长而总体呈增加趋势,堆制88 d时达到最高值,为1 g 8.841×106拷贝,而CK和T2处理该基因的表达量整体呈降低趋势。EC基因、CbhB基因表达量的变化趋势相似,均在中药渣堆制中后期开始有所增加,堆制88 d时,T1处理EC基因表达量达到最大值,为1 g 1.811×106拷贝;堆制65 d时,T2处理EC基因表达量最高,为1 g 1.592×106拷贝。在堆制65 d时,T1处理CbhB基因表达量最高,为1 g 5.720×1012拷贝。T1处理α62A基因表达量较高,且在堆制初期其表达量就有明显增加,在堆制46 d时其表达量达到最大值,为1 g 1.070×108拷贝。CK、T1处理和T2处理堆制46 d后,ChiA基因表达量明显增加,堆制65 d时T2处理ChiA基因表达量为1 g 6.800×108拷贝,高于T1处理,但在堆制88 d时T1处理ChiA基因表达量达到3个处理的最大值,为1 g 8.22×108拷贝。在堆肥过程中BLII基因表达量的变化趋势整体表现为先升高后降低,堆制46 d时,T1处理BLII基因表达量最高,为1 g 1.841×106拷贝。中药渣堆制65 d后,CK、T1处理、T2处理CP基因表达量均总体呈升高趋势,并且在堆制88 d时T1处理的表达量最高(1 g 3.310×107拷贝),其次为CK,T2处理的表达量相对较低。在中药渣堆肥期间,TI、T2处理Endo基因的表达量均总体呈升高趋势,堆肥88 d时T1处理Endo基因的表达量达到最大值,为1 g 2.270×1012拷贝。
3讨论
本研究以中药渣堆肥为研究对象,通过对不同时期样品中微生物群落结构变化以及腐熟过程中纤维素降解酶基因表达丰度的变化进行测定,探究中药渣堆制初期、中期、后期微生物群落结构、纤维素降解酶基因表达的动态变化特征。通过对中药渣堆肥温度进行记录可知,3个处理的中药渣堆肥在堆制46 d时均已进入高温期(温度≥55 ℃),随后高温一直持续至堆肥后期(88 d)。有研究结果表明,纤维素和木质素的降解主要发生在高温期和腐熟期[22]。因此,本研究的中药渣堆肥具有较长的高温腐熟期,可能与其含有大量难降解的纤维素和木质素密切相关。
Miseq高通量测序结果表明,厚壁菌门(Firmicutes)、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)和放线菌门(Actinobacteria)为中药渣堆肥在门分类水平上的优势菌群。醋酸杆菌属(Acetobacter)为属分类水平上的优势菌群,但随着堆制时间延长其所占比例总体呈减少趋势。假黄单胞菌属(Pseudoxanthomonas)存在于T1、T2处理中,魏瑟拉属(Weissella)存在于T1处理中,随着堆制时间延长其占比逐渐增加。目前,有很多研究结果证实假黄单胞菌属的细菌在纤维素降解中发挥着重要作用。例如,王志方等[23]发现与棉秸秆腐解有关的优势细菌菌属包括假黄单胞菌属(Pseudoxanthomonas),且其功能预测为降解纤维素、木质素、果胶类物质,提供氮素营养。侯丽媛等[24]发现假黄单胞菌菌株Pseudoxanthoonas sp.J1J1具有木质纤维素降解能力,该菌株具有内切纤维素酶(CMCase)、滤纸酶(FPase)和β-葡萄糖苷酶(BG)。同时,马宁等[25]发现一株纤维素高效降解细菌ZJ08,属于假黄单胞菌属,是一种具有较强耐酸特性的纤维素降解菌株。上述研究结果均表明假黃单胞菌属在纤维素降解中具有重要的作用。
通过对中药渣堆肥中微生物群落进行主成分分析(PCoA),发现随着中药渣堆制时间的延长,CK、T1处理、T2处理中微生物群落特征发生明显变化。同时,在堆肥末期,3个处理间群落结构差异更为明显,但3个组内的群落结构更趋于稳定。这说明,随着中药渣腐熟降解进程的推进,不同处理间微生物群落结构差异较大,微生物优势菌群也发生明显变化。同时,这种改变使不同处理的组内群落结构逐渐趋于稳定。艾士奇等[15]对复合菌系降解纤维素过程中微生物群落结构变化进行研究,发现在同一组别中,微生物群落特征随着时间的变化也发生着明显改变,不同降解时期的微生物群落分别分布在不同象限中,且相互之间存在一定的距离,表明在纤维素降解的不同时期,其复合菌系的微生物群落结构具有差异性。
本研究对中药渣堆制过程中纤维素降解酶基因的表达量变化也进行了检测。参与降解纤维素的微生物菌群可以分泌外切葡聚糖酶、内切葡聚糖酶和β-葡萄糖苷酶[26]等,它们协同降解纤维素大分子[27]。目前被广泛认可的纤维素降解理论为:首先,内切酶在纤维素分子的无定形区对β-1,4糖苷键进行随机切断,使长链纤维素产生大量反应末端。其次,外切酶作用于纤维素的结晶区,依次从纤维素分子末端切下纤维二糖或葡萄糖[28],这样在内切酶和外切酶的协同作用下,释放出聚合度很低的可溶性糖[29]。最后,由β-葡萄糖苷酶将纤维二糖等水解为葡萄糖。通过几种酶的协同作用可以彻底将聚合度高、分子量大的纤维素降解为溶于水的葡萄糖[30]。本研究在中药渣堆肥中选取了8种纤维素降解酶,包括:纤维素酶(AH18)[31]、纤维素水解酶(EC)[32]、纤维素分解酶(CbhB)[33]、阿拉伯糖苷酶(α62A)、几丁质内切酶(ChiA)、多糖基水解酶(BLII)、纤维二糖磷酸化酶(CP)、内切葡聚糖酶(Endo)。T1处理AH18基因表达量明显高于CK和T2处理,并随着堆制时间的延长而总体呈增加趋势,堆制88 d时达到最高值。纤维素酶在纤维素刚降解时就已经开始发挥作用了,该酶基因的高表达量一直持续到堆肥末期,表明纤维素酶在整个纤维素分解过程中均发挥着重要作用,具有纤维素酶基因的微生物菌群应该为中药渣堆肥中始终存在的微生物菌群。EC基因和CbhB基因的表达量变化趋势相似,均在中药渣堆制中后期开始有所增加,说明具有这2种酶基因的微生物菌群在堆肥初期发挥的降解作用不明显,直至堆肥的中后期,具有这2种酶基因的微生物菌群在整个微生物菌群中所占比例增加,而此时中药渣堆肥中仍含有大量未降解的纤维素物质,从而发挥重要的降解作用。
阿拉伯糖苷酶(α62A)和多糖基水解酶(BLII)均为可分解多糖的降解酶类。本研究中,T1处理的阿拉伯糖苷酶基因表达量在堆肥初期就有明显增加,在堆制46 d时其表达量达到最大值。多糖基水解酶基因在T1处理中表达量的变化趋势与阿拉伯糖苷酶基因类似,均为先缓慢升高,在堆肥中期(46 d)达到最大值后开始降低。此类多糖基水解酶基因表达量的变化趋势可解释为在堆肥初期大量纤维素类物质刚刚开始降解,此时起主要作用的是纤维素酶,将其初步分解产生大量的纤维素末端或降解为多糖类物质,随着堆肥的进一步腐熟,伴随纤维素的初步降解,大量多糖类物质被阿拉伯糖苷酶或多糖基水解酶降解,此时堆肥已经进入腐熟中期,其内存在大量具有阿拉伯糖苷酶或多糖基水解酶的微生物,可对堆肥初期已经分解的纤维素物质进行进一步分解。
纤维二糖磷酸化酶[34]和内切葡聚糖酶[35]均为可将多糖彻底降解为葡萄糖的重要酶类,其基因的表达在本研究中表现为:T1处理CP基因在堆肥中后期的表达量逐渐升高,至堆肥末期(88 d)达到最高值。与之类似,T1处理Endo基因表达量在堆肥中后期开始明显增加,在堆肥末期(88 d)达到最高值。说明,这2种酶基因表达量的大幅度增加是基于堆肥前期到中期大量的纤维素类物质分解为多糖类物质的基础上。随着中药渣堆肥的不断腐熟和降解,中后期存在大量具有阿拉伯糖苷酶或多糖基水解酶的微生物,可将多糖继续降解为分子量更小的纤维二糖或多聚糖,而随着中药渣堆肥腐熟度的进一步提高,堆肥末期具有纤维二糖磷酸化酶和内切葡聚糖酶的微生物菌群则开始成为优势菌群,发挥重要的分解作用,因此这2种降解酶类的基因表达量在堆肥末期最大。
4结论
本研究对中药渣堆肥样品进行Miseq高通量测序,检测其内微生物群落组成的变化。同时,对在中药渣堆肥腐熟过程中参与纤维素降解的重要降解酶基因的表达丰度也进行了探究。结果表明,随着中药渣堆肥中纤维素类物质的腐熟降解,其内微生物群落结构发生明显变化,具有纤维素降解功能的酶基因的表达量也发生相应变化,该变化随着堆制时间和堆肥温度的变化而不同。由此说明,中药渣堆肥中具有纤维素降解功能的微生物群落之间是相互联系、相互作用的。
参考文献:
[1]杨冰,丁斐,李伟东,等.中药渣综合利用研究进展及生态化综合利用模式[J]. 中草药,2017,48(2):377-383.
[2]贺灵芝,彭栋梁,梁忠厚. 中药渣开发利用探讨[J]. 科技创新与生产力, 2021(333):42-44.
[3]周曾艳,余高,陈芬,等. 添加畜禽粪便对中药渣好氧堆肥发酵特性及纤维素、木质素降解率的影响[J]. 河南农业科学, 2020,49(10):63-69.
[4]袁琪,李伟东,郑艳萍,等.中药渣的深加工及其资源化利用[J]. 生物加工过程,2019,17(2):171-176.
[5]王春芳,马诗淳,黄艳,等. 降解水稻秸秆的复合菌系及其微生物群落结构演替[J]. 微生物学报,2016, 56(12): 1856-1868.
[6]李璐. 高效降解中药渣菌群的筛选[D]. 杨凌:西北农林科技大学, 2019.
[7]毛莲英,李海碧,桂意云,等. 基于高通量测序分析间作猫豆对甘蔗根际土壤微生物的影响[J].南方农业学报,2021,52(2):332-340.
[8]孙瑞萍,王峰,晁哲,等. 1月龄五指山猪与长白猪骨骼肌miRNA转录组比较[J].江苏农业学报,2020,36(3):620-625.
[9]关健飞,曹阳.高通量测序分析黑土稀有微生物群落结构[J].江苏农业科学,2020,48(20):288-292.
[10]劳承英,韦本辉,周灵芝,等. 袋料种植淮山药根际土壤细菌多样性的高通量测序分析[J].南方农业学报,2020,51(10):2367-2373.
[11]陈娜,杨毅,张澜,等. Illumina 高通量测序技术分析早产儿出生后肠道菌群变化的初步研究[J]. 中国循证儿科杂志, 2014,9(5):359-364.
[12]BELLA J M D, BAO Y, GLOOR G B, et al. High throughput sequencing methods and analysis for microbiome research[J]. Journal of Microbiological Methods,2013,95(3):401-414.
[13]范海青,王凌文,王丹,等. 基于高通量测序的人工湿地微生物群落分析[J]. 科技通报,2019,35(2):213-219.
[14]胡丹丹,郭婷婷,李娜,等. 以16S rDNA高通量測序法研究果寡糖对奶牛瘤胃菌群结构及多样性的影响[J]. 中国畜牧兽医,2018,45 (10):2761-2771.
[15]艾士奇,赵一全,孙志远,等. 复合菌系降解纤维素过程中微生物群落结构的变化[J]. 生物工程学报,2018,34(11):1794-1808.
[16]李淼淼,沈飞,张庆华.纤维素降解复合菌系的微生物多样性及关键功能菌解析[J].环境科学学报,2018,38(7):2690-2695.
[17]CAPORASO J G, LAUBER C L, WALTERS W A, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample [J].Proceedings of the National Academy of Sciences,2011,108:4516-4522.
[18]MENG T Z, YANG Y J, CAI Z C, et al. The control of Fusarium oxysporum in soil treated with organic material under anaerobic condition is affected by liming and sulfate content [J]. Biology and Fertility of Soils,2018,54:295-307.
[19]CAPORASO J G, KUCZYNSKI J, STOMBAUGH J, et al. QIIME allows analysis of high-throughput community sequencing data [J]. Nat Methods,2010,7:335-336.
[20]SHEN Z Z, RUAN Y Z, WANG B B, et al. Effect of biofertilizer for suppressing Fusarium wilt disease of banana as well as enhancing microbial and chemical properties of soil under greenhouse trial[J]. Applied Soil Ecology,2015,93:111-119.
[21]LIU J F, LIU H Y, QI F S, et al. Application of nano-TiO2/chitosan composite preservative on Penaeus vannamei preservation [J]. Food Science and Technology,2014,39(2):245-249.
[22]邓辉,王成,吕豪豪,等. 堆肥过程放线菌演替及其木质纤维素降解研究进展[J]. 应用与环境生物学报,2013,19 (4):581-586.
[23]王志方,陈竞,代金平,等. 棉秸秆自然腐解过程中细菌菌群多样性分析[J]. 新疆农业科学,2019,56(1):117-125.
[24]侯丽媛,江经纬,蒋建东,等. 假黄单胞菌株J1的筛选及木质纤维素降解基因的生物信息学分析[J]. 南京农业大学学报,2016,39 (4):573-581.
[25]马宁,魏姜勉,张亮. 一株牛瘤胃耐酸纤维素降解细菌的筛选、鉴定及酶学特性分析[J]. 湖北畜牧兽医,2015,36(6):7-8.
[26]LYND L R, WEIMER P J, VANZYL W H, et al. Microbial cellulose utilization: fundamentals and biotechnology[J]. Microbiology and Molecular Biology Reviews,2002,66(3):506-577.
[27]BRETHAUER S, STUDER M H. Biochemical conversion processes of lignocellulosic biomass to fuels and chemicals - a review [J]. Chimia International Journal for Chemistry,2015,69 (10):572-581.
[28]ZHANG Y H, LYND L R. Toward an aggregated understanding of enzymatic hydrolysis of cellulose: Noncomplexed cellulase systems [J]. Biotechnology and Bioengineering,2004,88 (7):797-824.
[29]SAWATDEENARUNAT C, SURENDRA K C, TAKARA D, et al. Anaerobic digestion of lignocellulosic biomass: challenges and opportunities [J]. Bioresource Technology,2014,178:178-186.
[30]MOHAMED A H, YOUSEIF S H, EL-MAGEED F H A, et al. Production of cellulase, exoglucanase and xylanase by different microorganisms cultivated on agricultural wastes[J]. Research Journal of Pharmaceutical Biological and Chemical Sciences,2017,8(4):435-452.
[31]LIM J H, AHN C H, JEONG H Y, et al. Genetic monitoring of multi-functional plant growth promoting rhizobacteria Bacillus subtilis AH18 and Bacillus licheniformis K11 by multiplex and real-time polymerase chain reaction in a pepper farming field[J]. Journal of the Korean Society for Applied Biological Chemistry,2011,54 (2):221 -228.
[32]BUSSLER L, JACOMINI D, CORRA J M, et al. Recombinant cellulase of Caulobacter crescentus: potential applications for biofuels and textile industries[J].Cellulose,2021,28(5):2813-2832.
[33]MAHMOOD N, AHSAN M, AHMAD I, et al. Molecular expression profile of different cellulolytic enzyme genes in Aspergillus niger in response to UV radiation and chemical mutagenesis [J]. Cellulose Chemistry and Technology, 2014,48(5):5-6.
[34]UBIPARIP Z, MORENO D S, BEERENS K, et al. Engineering of cellobiose phosphorylase for the defined synthesis of cellotriose [J]. Applied Microbiology and Biotechnology,2020,104 (19):8327-8337.
[35]HAYASHI T, WONG Y S, MACLACHLAN G. Pea xyloglucan and cellulose: II. hydrolysis by pea endo-1,4-β-glucanases [J]. Plant Physiology,1984,75(3):605-610.
(責任编辑:王妮)
收稿日期:2021-08-27
基金项目:国家重点研发计划项目(2017YFD0800200);江苏省农业科技自主创新基金项目[CX(17)2025];中国博士后科学基金项目(2018M632255);江苏农林职业技术学院青年支持项目(2020kj011)
作者简介:杜婕(1985-),女,山东泰安人,博士,讲师,主要从事降解功能微生物研究。(E-mail)dujiekycg@163.com
通讯作者:马艳,(E-mail)myjaas@sina.com
猜你喜欢
热带农业科学(2016年11期)2017-01-21
热带农业科学(2016年11期)2017-01-21
河北渔业(2015年12期)2015-12-21
河北渔业(2015年11期)2015-11-18
河北渔业(2015年11期)2015-11-18
河北渔业(2015年10期)2015-10-15
河北渔业(2015年7期)2015-07-21
河北渔业(2015年6期)2015-06-24
安徽农学通报(2015年5期)2015-03-23
安徽农学通报(2015年5期)2015-03-23
