
Genetic diversity analysis and GWAS reveal the adaptive loci of milling and appearance quality of japonica rice (Oryza sativa L.) in Northeast China
2022-05-09 03:37XUXinYEJunhuaYANGYingyingLIRuosiLIZhenWANGShanSUNYanfeiZHANGMengchenXUQunFENGYueWEIXinghuaYANGYaolong
XU Xin,YE Jun-hua,YANG Ying-ying,LI Ruo-si,LI Zhen,WANG Shan,SUN Yan-fei,ZHANG Mengchen,XU Qun,FENG Yue,WEI Xing-hua,YANG Yao-long
State Key Laboratory of Rice Biology,China National Rice Research Institute,Hangzhou 311400,P.R.China
Abstract Milling and appearance quality are important contributors to rice grain quality. Abundant genetic diversity and a suitable environment are crucial for rice improvement. In this study,we investigated the milling and appearance qualityrelated traits in a panel of 200 japonica rice cultivars selected from Liaoning,Jilin and Heilongjiang provinces in Northeast China. Pedigree assessment and genetic diversity analysis indicated that cultivars from Jilin harbored the highest genetic diversity among the three geographic regions. An evaluation of grain quality indicated that cultivars from Liaoning showed superior milling quality,whereas cultivars from Heilongjiang tended to exhibit superior appearance quality.Single-and multi-locus genome-wide association studies (GWAS) were conducted to identify loci associated with milling and appearance quality-related traits. Ninety-nine significant single-nucleotide polymorphisms (SNPs) were detected.Three common SNPs were detected using the mixed linear model (MLM),mrMLM,and FASTmrMLM methods. Linkage disequilibrium decay was estimated and indicated three candidate regions (qBRR-1,qBRR-9 and qDEC-3) for further candidate gene analysis. More than 300 genes were located in these candidate regions. Gene Ontology (GO) analysis was performed to discover the potential candidate genes. Genetic diversity analysis of the candidate regions revealed that qBRR-9 may have been subject to strong selection during breeding. These results provide information that will be valuable for the improvement of grain quality in rice breeding.
Kewwords:rice,grain quality,GWAS,genetic diversity
1.Introduction
Rice is an important staple food crop that has a long history of cultivation. High yield and grain quality are the primary goals of rice breeding. During artificial selection and natural evolution,rice populations have gradually adapted to the local climate and environment,and they have also been subjected to selection to suit human consumption preferences. These processes have led to the increased frequency of dominant alleles that control target traits or alleles located on chromosome segments,and the decreased polymorphism of chromosomal sequences linked with a target gene.
Northeast China is an importantjaponicarice-growing area in China that is renowned for the high quality of grains produced in the region (Xinet al.2020). The unique climate of the region plays an important role in determining grain quality. During the rice-growing season in Northeast China,the daily maximum temperature,daily minimum temperature,and accumulated temperature gradually decrease from the southwest to the northeast(Shiet al.2020). The overall solar radiation is high in the south and low in the north. Furthermore,total precipitation gradually decreases from the southeast to the northwest.In addition,previous reports indicate that rice cultivars grown in the three provinces of Northeast China differ in their overall quality traits;rice produced in Heilongjiang Province usually exhibits good eating qualities,whereas cultivars grown in Liaoning Province typically produce high yields (Xuet al.2006;Liet al.2013).
Rice grain quality is closely linked with consumer preferences. Grain quality,especially the milling quality,is the preferable index of rice yield,rather than the rice paddy yield,because the grain quality is impacted during the milling process (Misraet al.2019). In addition,grain quality is a determinant of economic value and grain quality preferences typically vary among consumers in different regions. In general,rice grain quality includes the milling quality and exterior quality,as well as the eating and cooking quality (Sharet al.2020). The milling quality comprises the brown rice recovery,milled rice recovery,and head rice recovery. The exterior quality includes the grain shape,endosperm translucency,percentage of chalkiness,and degree of chalkiness. The cooking and eating quality includes the gelatinization temperature,gel consistency,and amylose content (Qiuet al.2015).Among these attributes,the milling and appearance quality directly influence the yield and commodity value.Given that Northeast China is an important rice production region (Weiet al.2008;Cuiet al.2020),it is crucial to analyze the genetic basis of quality traits among the rice cultivars currently grown in this region so that cultivars with improved grain quality can be bred.
Many studies of rice milling quality have been reported.Milling quality is a quantitative trait controlled by many quantitative trait loci (QTLs) and may be influenced by grain shape (Tanet al.2001). Milling quality QTLs overlap with the major genes that determine grain shape. In addition,the milling quality is influenced by grain chalkiness. If endosperm starch and protein accumulation is impaired,the grain is readily damaged during processing. Grain chalkiness is also a quantitative trait that is regulated by major QTLs. This trait is closely associated with source-sink regulation. Temperature is an additional crucial factor that determines grain chalkiness(Lanninget al.2011;Siebenmorgenet al.2013).
To date,a number of major QTLs for rice milling quality have been identified,but no genes have been cloned and functionally characterized. However,several QTLs associated with chalkiness have been cloned,such asChalk5(Li Yet al.2014),SMALLKERNEL1(OsSMK1) (Li X Jet al.2014),GLUTELINPRECURSOR ACCUMULATION4(GPA4/GOT1B) (Wang Yet al.2016),SUBSTANDARDSTARCHGRAIN4(SSG4) (Matsushimaet al.2014),andFLOURYENDOSPERM4(FLO4/OsPPDKB) (Kanget al.2005).Chalk5as a major QTL involved in regulating the percentage of grain chalkiness that has been cloned (Li Yet al.2014),and it also affects rice head rice yield and many other quality traits.Chalk5encodes a vacuolar H+-translocating pyrophosphatase that couples inorganic pyrophosphate hydrolysis and H+-translocation activity. Plants that over-expressChalk5display increased chalkiness of the endosperm.GPA4encodes a protein that mediates the formation of coat protein complex II vesicles at endoplasmic reticulum exit sites,thereby facilitating anterograde transport of the secretory proteins in plant cells. Mutants show abnormally high concentrations of endosperm 57 kD glutelin precursors and the luminal chaperone binding protein,which results in a chalky or silty quality of the grain (Fukudaet al.2016;Wang Yet al.2016).
Genome-wide association study (GWAS) has been widely used to analyze the genetic basis of complex agronomic traits in various crops,such as maize,rice,wheat,and soybean (Wanget al.2018). Based on phenotype and genotype data,GWAS is an effective means for isolating the gene associated with a target trait.For example,in rice,GRAINLENGTHANDWEIGHTON CHROMOSOME7(GLW7/OsSPL13) (Siet al.2016),GRAINWIDTH5(GW5/GSE5) (Duanet al.2017),andqPSR10(Xiaoet al.2018) have been identified by GWAS.In the present research,we combined GWAS and population compositional analysis to identify candidate regions involved in the regulation of milling quality and appearance,which can be selected during breeding to improve grain quality.
2.Materials and methods
2.1.Plant materials
A panel comprising 200japonicarice cultivars grown in three provinces of Northeast China was used in this study. The panel included 57 cultivars from Heilongjiang Province,22 cultivars from Jilin Province,and 121 cultivars from Liaoning Province. The cultivars were selected from a previously reported population (Yeet al.2018). Plants were grown in the experimental field in Shenyang (41°48´N,123°25´E),Liaoning Province. Each accession was grown in plots of 20 cm×20 cm,with each plot comprising four rows with six individuals per row,during the summer of 2016. After harvest,the airdried grains were collected from each line for phenotype evaluation.
2.2.Phenotype evaluation
A total of 30 g of grains that were thoroughly dried for approximately 3 months were dehulled using an electrical dehuller (JLGJ4.5,Xinfeng,China) and milled using a sample miller (Model JB-20,China). Head rice that had been dehulled for each accession was used to measure the degree of endosperm transparency (DET),degree of endosperm chalkiness (DEC),and percentage of grains showing chalkiness (PGC). In accordance with theChina National Rice Grain Quality Assessment Standard(NY/T 593-2013 2013),the traits associated with milling quality were assessed,namely brown rice recovery (BRR),milled rice recovery (MRR),and head rice recovery (HRR).All measurements for each accession were conducted with two replications. In addition,the milling quality,appearance quality,and eating quality of grains from each cultivar were assessed. The cultivars comprised five second-class accessions,69 third-class accessions,and 126 ordinary-class accessions.
2.3.Genotyping
Single-nucleotide polymorphism (SNP) genotype data for the 200 accessions were derived from a 90 K high-density SNP array. After removal of the SNPs with minor allele frequency <0.05 and minimum count <90% of the 200 accessions using TASSEL 5.2.51 (Bradburyet al.2007),23 695 SNPs were selected for GWAS.
2.4.Data analysis
Statistical analysis of grain quality-related traits was conducted using Microsoft Excel 2010. Analysis of variance andposthocmultiple comparisons were conducted with SAS 9.2 and R 4.0.0. Correlation analysis of the grain quality-related traits was performed using R.Principal component analysis (PCA) of these traits was performed with R based on the correlation matrix between the traits,and the principal components (PCs) that collectively explained more than 90% of the phenotypic variation were selected. Genetic diversity parameters,includingNei’s genetic distance andF-statistics (Fst),were calculated using Powermarker 3.25. Sequence diversity(π) and linkage disequilibrium (LD) decay distance were calculated using TASSEL 5.2.51. A neighbor-joining(NJ) phylogenetic tree was generated with MEGA 6.0 Software.
2.5.Genome-wide association study
A mixed linear model (MLM) was used for GWAS using kinship and PC matrices as covariates to correct for population structure. Calculation of the kinship and PC matrices was conducted using TASSEL 5.2.51. In addition,a multi-locus GWAS (Wenet al.2018) was performed to verify the peaks identified by the fixed-SNP-effect MLM GWAS. The Bonferroni-corrected threshold for theP-value was 10/23 695 (P=α/n,α=10).For simplicity,P<0.0004 was used as the threshold value.The FASTmrMLM (Tamba and Zhang 2018) and mrMLM(Wang S Bet al.2016) algorithms implemented in R were used to conduct the multi-locus GWAS.
2.6.Candidate gene analysis
On the basis of the GWAS results,we performed LD analysis to discover the candidate regions for the significant loci using Haploview Software (Barrettet al.2005). The AgriGO v2 database (http://bioinfo.cau.edu.cn/agriGO/analysis.php) was used to conduct a Gene Ontology (GO) enrichment analysis. The expression patterns of genes in the candidate region were obtained from the RNA-seq database of the Rice Genome Annotation Project (http://rice.plantbiology.msu.edu/).
3.Results
3.1.Clustering analysis of cultivars with different geographic distributions
With consideration of the provenance of the rice cultivars(Appendix A) and SNP genotypes,a NJ tree was constructed based on theNei’s genetic distances among the cultivars (Fig.1-A). The cultivars from Heilongjiang and Liaoning formed separate clusters,whereas cultivars from Jilin were mixed among those from the other two provinces. Similarly,PCA resolved the cultivars from Heilongjiang and Liaoning into two distinct groups,and the cultivars from Jilin were mixed among these two groups(Fig.1-B and C). The highest pairwiseFst-value between the Heilongjiang and Liaoning cultivars was 0.1705,whereas theFst-values between either the Heilongjiang and Jilin cultivars,or the Liaoning and Jilin cultivars were comparatively low. These results indicated that cultivars from Jilin showed a relatively close genetic relatedness with cultivars from both Heilongjiang and Liaoning.
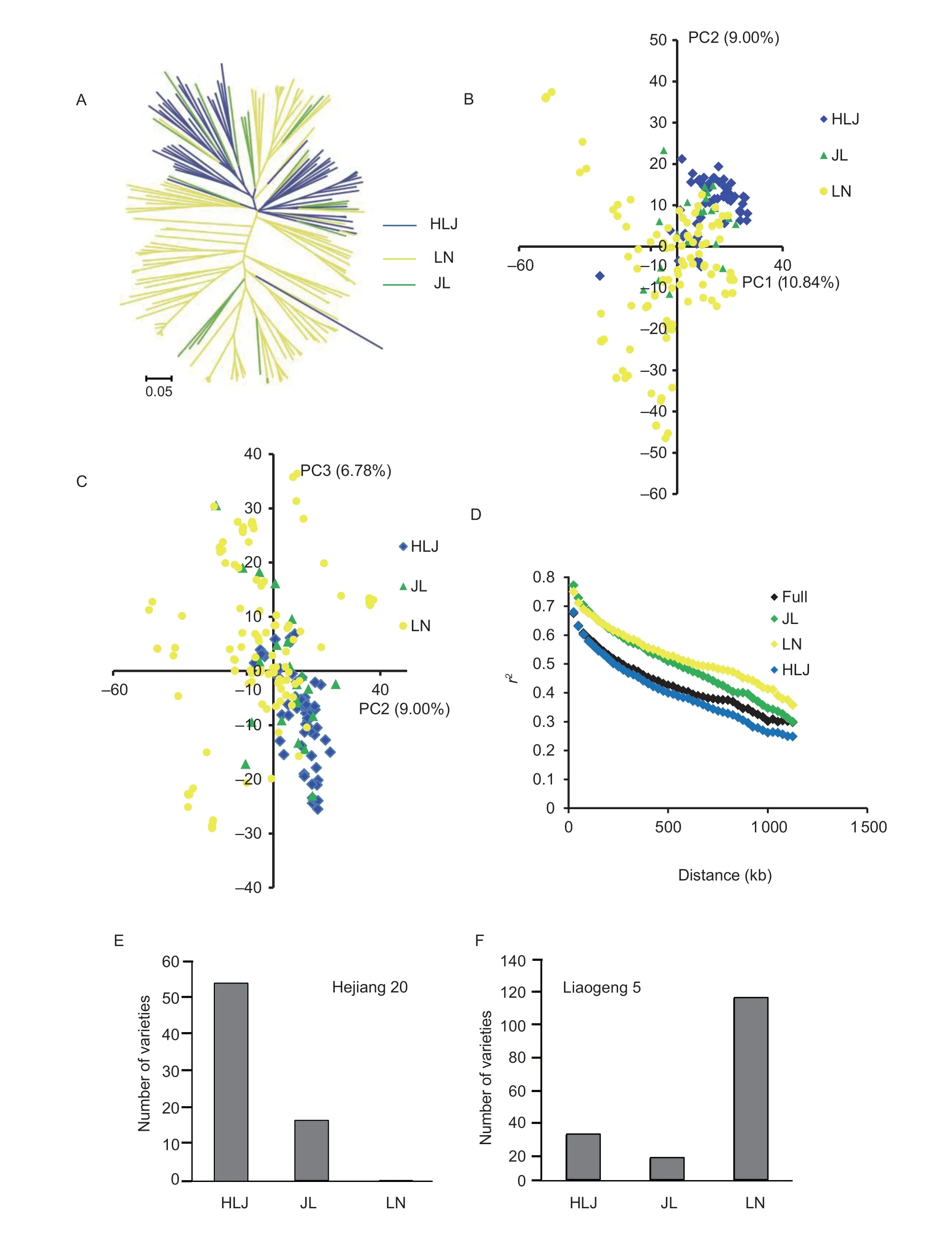
Fig.1 Genetic analysis and pedigree analysis of the rice cultivars in Northeast China. A,unrooted neighbor-joining tree. B,principal component analysis (PCA) plots of the principal component 1 (PC1) and PC2 of 200 cultivars. C,PCA plots of the PC2 and PC3 of 200 cultivars. D,genome-wide average linkage disequilibrium (LD) decay values estimated from Liaoning (LN),Heilongjiang(HLJ),Jilin (JL) and full groups. E,the number of varieties which used Hejiang 20 as the parents in the three provinces. F,the number of varieties which used the Liaogeng 5 as the parents in the three provinces.
To investigate the genetic diversity ofjaponicarice cultivars in relation to their provenance,we calculated the genome-wide LD decay distance and π for the cultivars from each province. The LD analysis revealed that the Liaoning group showed the highest LD decay distance(Fig.1-D). The average π value was estimated for each province according to the SNP genotypes. The cultivars from Jilin showed the highest π value (0.00036728),whereas the cultivars from Liaoning exhibited the lowest π value (0.00030693). The analysis of the overall genetic variation among the cultivars in the three groups was performed and yielded similar results. Thus,the cultivars from Liaoning Province showed the lowest genetic diversity among the cultivars analyzed (Appendix B).
Given the distinct clustering of cultivars from Heilongjiang and Liaoning provinces in the NJ tree,together with the strong differentiation of subpopulations within each cluster,we investigated the pedigree of the cultivars. The cultivar ‘Liaogeng 5’ was bred in Liaoning Province and had been widely used as a parent in both Heilongjiang and Jilin provinces. ‘Hejiang 20’ was bred in Heilongjiang Province and was widely used as a parent in both Heilongjiang and Jilin provinces,but not for breeding in Liaoning Province (Fig.1-E and F). These observations were consistent with the finding that cultivars from Heilongjiang and Liaoning had the highestFst-values.
3.2.Phenotypic evaluation
In general,the milling quality-related traits (BRR,MRR,and HRR) and appearance quality-related traits (DEC,DET,and PGC) showed relatively uniform frequency distributions (Table 1;Fig.2-A). However,DET showed a distinctly skewed distribution and lower variation.Correlation analysis revealed that the three milling quality-related traits were positively correlated,and thethree appearance quality-related traits were positively correlated. However,no significant correlation between the milling-and appearance-quality traits was observed,and only PGC showed a weak positive correlation with BRR (Fig.2-B). In addition,comparison of the six grainquality traits among the cultivars from the three provinces revealed that cultivars from Liaoning showed superior milling quality compared with those of cultivars from the other two provinces. With regard to appearance qualityrelated traits,significant differences were observed between the Heilongjiang and Liaoning provinces,which indicated that the appearance quality of cultivars from Heilongjiang tended to be superior to that of cultivars from Liaoning (Appendix C).

Table 1 Phenotypic variation of all cultivars
Given the significant correlations observed among the quality-related traits,PCA was performed for the six traits to determine the most important factors underlying the phenotypic variation in the study population. The first and second PCs explained 75.9% of the total phenotypic variation in the population (Appendix D). All six traits showed negative loadings on PC1 and the milling qualityrelated traits showed higher loadings. With regard to PC2,milling quality-related traits showed positive loadings but appearance quality-related traits showed negative loadings (Appendix D),and the loadings of appearance quality-related traits were higher. PC1 and PC2 each displayed a continuous distribution and that of the PC1 was similar to a normal distribution (Fig.2-C). In conjunction with the evaluation of grain quality of these cultivars (Appendix A),most of the second-and thirdclass cultivars showed a low PC1 loading and a high PC2 loading. In contrast,most ordinary-class cultivars showed a high PC1 loading and a low PC2 loading (Fig.2-D).These results indicated that the PCs well-represented the milling quality and appearance quality of the cultivars in the study population.

Fig.2 Phenotypic evaluation of the rice cultivars in Northeast China. A,distribution of milling and appearance quality traits in the japonica varieties of Northeast China. B,correlation analysis of brown rice recovery (BRR),milled rice recovery (MRR),head rice recovery (HRR),degree of endosperm chalkiness (DEC),degree of endosperm transparency (DET) and percentage of grains showing chalkiness (PGC).,no significant correlation. C,distribution of the principal component 1 (PC1) and PC2 for the six milling and appearance quality traits. D,phenotypic principal component analysis (PCA) plots of the PC1 and PC2 of the 200 cultivars.
3.3.GWAS for PCs and traits
The kinship matrix and PC1-PC3 were used as covariates to perform association mapping. In a single-locus GWAS(MLM),40 SNPs that passed the filtering threshold were detected on chromosomes 1,2,3,8,9,10,and 12. To verify the peaks identified by the single-locus GWAS(Table 2),we also conducted a multi-locus GWAS (mrMLM and FASTmrMLM). As a result,26 of the suggestive SNPs distributed on chromosomes 1,2,3,4,5,6,8,9,and 11 were detected using the mrMLM method and 23 of the suggestive SNPs were identified by the FASTmrMLM method. Three SNPs were co-detected with the singlelocus and multi-locus GWAS approaches (Appendix E).
In the single-locus GWAS,11 SNPs were detected for milling quality-related traits and 29 SNPs were detected for appearance quality-related traits (Table 2). For milling quality-related traits,one SNP (seq9_6319797) was detected that accounted for 7.03% of the phenotypic variation explained by PC1 (Fig.3). This SNP was also detected for BRR and MRR,which accounted for 10.24 and 9.17% of the phenotypic variation,respectively. A single SNP,seq2_10801110,was detected for HRR that explained 8.40% of the phenotypic variation. Among the appearance quality-related traits,no SNP was detectedfor DET. The SNP Seq3_28028330 was detected for both DEC and PC2 (Fig.4),and accounted for 8.58 and 6.90% of the phenotypic variation,respectively (Table 2;Appendix E). In addition,an SNP-dense region on chromosome 2 associated with DEC and PGC was detected;the most important SNP,seq2_17858477,accounted for 8.25 and 7.67% of the phenotypic variation in DEC and PGC,respectively (Table 2;Appendix E).
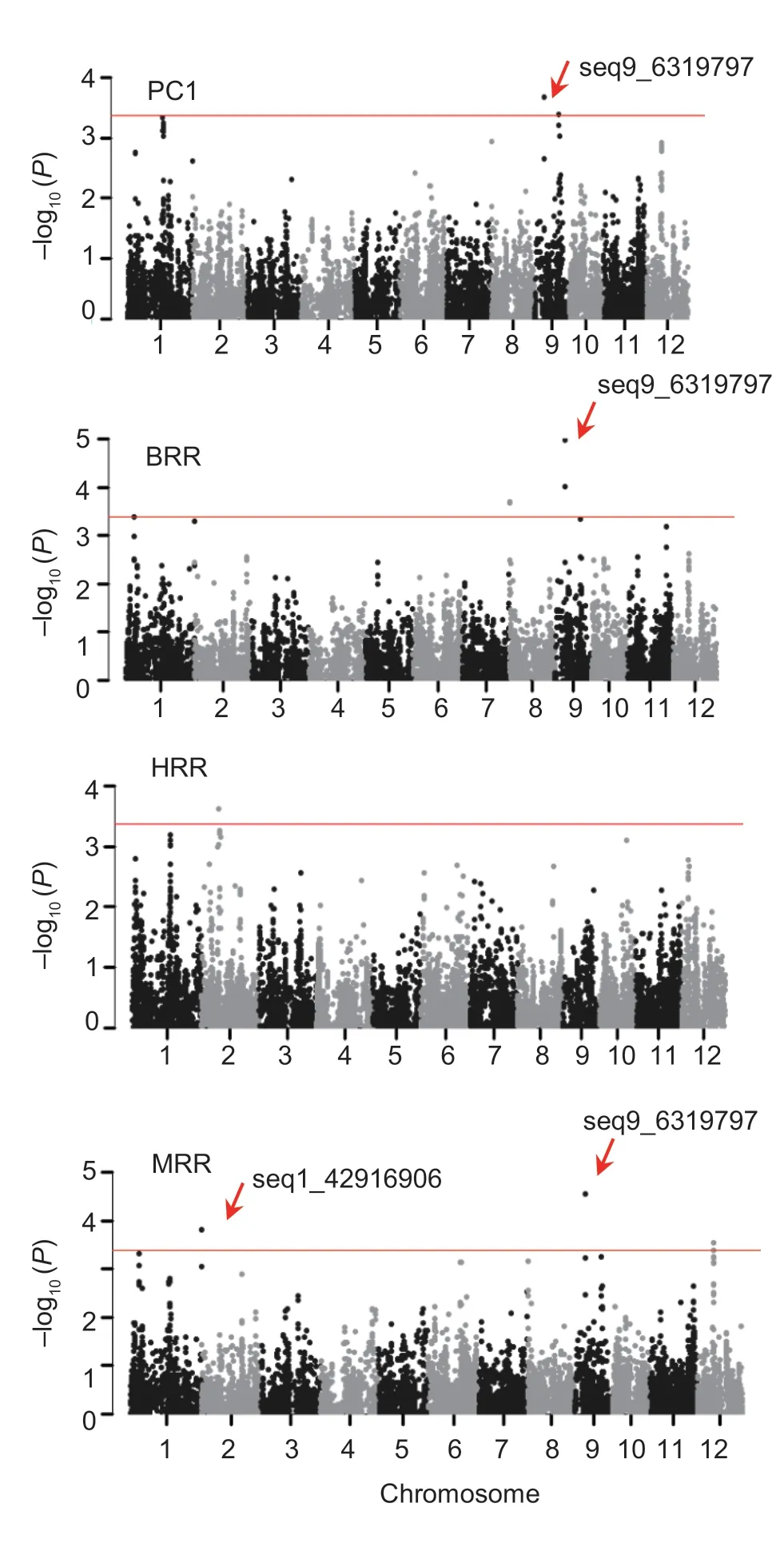
Fig.3 Genome-wide association studies of principal component 1(PC1),brown rice recovery (BRR),milled rice recovery (MRR),and head rice recovery (HRR).

Fig.4 Genome-wide association studies of principal component 2(PC2),degree of endosperm chalkiness (DEC) and percentage of grains showing chalkiness (PGC).

Table 2 Summary of the significant SNPs detected by single-locus genome-wide association studies (GWAS)
In the multi-locus GWAS,11 SNPs associated with milling quality-related traits and 15 SNPs associated with appearance quality-related traits were detected by the mrMLM method,whereas 8 and 15 SNPs were detected using FASTmrMLM,respectively. Among these SNPs for milling quality-related traits,no SNP was detected for HRR. The SNP seq9_6319797 showed the highest LOD scores for MRR,BRR,and PC1 in the mrMLM Model.For the FASTmrMLM Model,seq1_42916906 showed the highest LOD score for PC1,seq8_51045 showed the highest LOD score for BRR,and seq9_6319797 had the highest LOD score for MRR. For appearance quality-related traits,seq3_28028330 showed the highest LOD scores for DEC and PC2 in both mrMLM and FASTmrMLM models,and was also detected for PGC with the FASTmrMLM Method. The SNP seq4_3407655 was detected for DET in the mrMLM and FASTmrMLM models.
Among the suggestive SNPs,three SNPs were detected by all three GWAS methods. The SNPs seq1_42916906 and seq9_6319797 were associated with milling quality-related traits,and seq3_28028330 was associated with appearance quality-related traits.For seq1_42916906,the GG genotype had a positive effect on PC1 and is undesirable for rice milling quality.For seq9_6319797,the TT genotype had positive effects on BRR and MRR,and a negative effect on PC1. For seq3_28028330,the AA genotype had a positive effect on PC2,and negative effects on DEC and PGC. On the basis of these results,we analyzed the utilization of the three elite SNP genotypes in the three provinces.Seq1_42916906-TT and seq9_6319797-TT were detected in almost all cultivars bred in Liaoning,but were rarely detected in cultivars bred in Heilongjiang (Appendix F). This observation may account for the higher BRR of cultivars from Liaoning than cultivars from Heilongjiang(Appendix G). Seq3_28028330-AA was detected in the majority of cultivars from all three provinces,although the number of cultivars harboring the AA genotype in Heilongjiang was fewer than in Liaoning. However,the proportion of cultivars carrying seq3_28028330-AA exceeded those harboring seq1_42916906-TT or seq9_6319797-TT.
3.4.Candidate gene analysis
According to our previous research,the LD distance of this population is approximately 1 Mb. Therefore,the SNPs within 1 Mb of the suggestive SNPs detected in the present study were used for LD analysis to identify the candidate region. Three candidate regions were identified:qBRR-1,qBRR-9,andqDEC-3(Appendix H).The candidate regionqBRR-1was localized to the interval 42.8-42.9 Mb (59 kb) on chromosome 1 and contained 16 genes according to the Rice Genome Annotation Project Database (RAP-DB) (Appendix I). The candidate regionqBRR-9was located in the interval 58.6-68.1 Mb(492 kb) on chromosome 9 and was annotated with 118 genes in the RAP-DB. The candidate regionqDEC-3was localized to 27.4-28.7 Mb (1 403 kb) on chromosome 3 and contained more than 200 genes according to RAPDB (Appendix I). Analysis of such a large number of genes in the latter candidate region was not practical,so only the candidate genes inqBRR-1andqBRR-9were investigated in the following GO analysis.
First,we identified 21 cloned genes associated with the endosperm,starch synthesis,and chalkiness from the China Rice Data Center database (http://www.ricedata.cn/index.htm). These genes,together with the candidate genes identified in the association analysis,were used for GO analysis (Appendix J). Three genes located in theqBRR-9candidate region were annotated with putative functions and GO terms. Among these three genes,LOC_Os09g11460 and LOC_Os09g11480 encode an ethyleneresponse factor transcriptional regulator that shared the same functional annotation as the geneETHYLENE RESPONSEFACTOR131(ERF131/SERF1). The GO enrichment analysis revealed that these two genes were assigned the same GO terms,namely DNA-binding transcription factor activity,regulation of transcription(DNA-templated),and cellular macromolecule biosynthetic process. The gene LOC_Os09g10940 encodes a Rasrelated protein assigned with a putative function as a GTPbinding protein and was annotated with the same GO terms asSECRETION-ASSOCIATED,RAS-RELATED PROTEIN1A(OsSar1a),OsSar1b,andOsSar1c,namely small GTPase-mediated signal transduction,intracellular protein transport,intracellular anatomical structure,signaling,signal transmission,and GTP binding.
3.5.Evidence for selection at candidate regions
Given the differences in the distributions of elite SNPs among the three provinces,we inferred that the SNPs had been selected in different environments (Fig.5). The SNPs in the three candidate regions were used to calculate genetic diversity. Comparison of the genetic diversity among the three regions revealed that the Heilongjiang group showed higher diversity than the Liaoning group,especially in theqBRR-9region. This result may indicate that the candidate regions were under strong selective pressure in the cultivars bred in Liaoning.

Fig.5 Genetic diversity analysis of the three candidate regions. A,genetic diversity variation in qBRR-9. B,genetic diversity variation in qBRR-1. C,genetic diversity variation in qDEC-3. HLJ,Heilongjiang;JL,Jilin;LN,Liaoning. PIC,polymorphism information content.
4.Discussion
4.1.Genetic diversity of the rice cultivars in three provinces of Northeast China
In previous research,we concluded that the original parents of the rice cultivars bred in Northeast China were dominated by the cultivars ‘Liaogeng 5’,‘Liaogeng 326’,‘Songgeng 3’,and ‘Hejiang 20’ (Yanget al.2020).In the present study,we demonstrated that the original parents utilized differed in each of the three provinces.Construction of a NJ tree indicated that the cultivars from Jilin showed a mixed relationship with the cultivar groups from Heilongjiang and Liaoning,and that the cultivars from Heilongjiang and Liaoning formed distinct clusters.The degree of genetic differentiation between the Jilin population and the other two groups was low,whereas the genetic differentiation between the Heilongjiang and Liaoning populations was relatively high. A similar result was reported previously (Liuet al.2015). The breeding pedigree of the cultivars aids in interpreting these results.Liaogeng 5 and Hejiang 20,two of the predominant original parents,were bred in Liaoning and Heilongjiang,respectively. Liaogeng 5 was used successfully in cultivar improvement in all three provinces,whereas Hejiang 20 was only utilized in Heilongjiang and Jilin. This difference may be explained by the differences in temperature between Heilongjiang and Liaoning. When a cultivar is introduced from the north to the south,the growth period is often shortened because of the higher temperature in the south. Thus,Hejiang 20 was not utilized for breeding in Liaoning. The analysis of genetic diversity showed that the cultivars from Jilin harbored higher genetic diversity than the cultivars from the other two provinces.In addition,the cultivars from Heilongjiang showed lower LD decay distances than those of cultivars from Jilin.These results may be expected because cultivars from Liaoning and Heilongjiang were used as breeding parents for the cultivars from Jilin,which also explains the mixed relationships of the Jilin subpopulation.
4.2.Variation in grain quality-related traits in the study population
The present study population comprised improved rice cultivars from Northeast China. The improved cultivars are of high economic value and exhibit high yield,good quality,or both traits (Xuet al.2016). This may account for the skewed phenotype distributions that were observed.In addition,appearance quality-related traits showed low variation compared with milling quality-related traits,which may reflect the fact that appearance quality is simpler to select than milling quality during breeding. Correlation analysis showed that all measured traits associated with milling quality or appearance quality were significant,which was consistent with previous findings (Wanget al.2017).The strong correlations between traits suggested that the analyzed traits were suitable for PCA and the phenotypic variation could be represented in few dimensions. The present results indicated that PC1 was an important negative factor for milling quality and PC2 was an important negative factor for appearance quality. In addition,the distributions of cultivars classified into different rice grain quality classes confirmed that second-and third-class cultivars usually show a low PC1 loading and a high PC2 loading,whereas ordinary cultivars commonly show a high PC1 loading and a low PC2 loading.
4.3.GWAS of rice grain quality
By performing single-and multi-locus GWAS analyses,we identified QTLs associated with milling quality and appearance quality. A small number of QTLs accounted for a large proportion of the phenotypic variation and many QTLs showed a lower marker interpretation rate,especially for milling quality. Thus,it is possible that many minor loci were responsible for the variation in milling quality-related traits in the study population.
The improved cultivars from Heilongjiang,Jilin and Liaoning exhibited differences in phenotypic traits. In addition to adaptation to local environments,this also likely reflects divergent breeding goals in the different regions. In combination with the geographical distribution of the cultivars,we observed differences in the spatial distribution of significant SNPs with major effects. The elite SNPs for milling quality were predominantly detected in cultivars from Liaoning and were rarely detected in Heilongjiang cultivars. This difference may be primarily associated with distinct breeding goals and corresponded with the superior milling quality of cultivars from Liaoning.Cultivars from Liaoning and Jilin showed reduced genetic diversity in theqBRR-9region,which likely reflected that this region was selected during breeding for high yield.Among cultivars from Heilongjiang,the candidate regionqBRR-1associated with milling quality showed high genetic diversity,whereas theqDEC-3region associated with appearance quality-related traits exhibited low genetic diversity. These results may reflect that high yield was the primary breeding goal in Liaoning. The present results indicated that the foundational parent cultivars used in rice breeding in each of the three provinces were introduced from the other two provinces. Therefore,the introduction of suitable unrelated cultivars in breeding programs would enhance the population genetic diversity in each province.
5.Conclusion
We observed variation in grain quality and the genetic backgrounds in the populations of rice cultivars from three provinces in Northeast China. This diversity may reflect differences in the selection goals during breeding. In addition,by conducting association analysis,we identified putative QTLs associated with milling quality and the linked significant SNPs,which may be useful for markerassisted selection in breeding.
AcknowledgementsThis research was funded by the National Key Research and Development Program of China (2016YFD0100902-07),the Central Public-interest Scientific Institution Basal Research Fund,China (CPSIBRF-CNRRI-202101),and the Chinese Academy of Agricultural Sciences (CAASASTIP-201X-CNRRI).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
 Journal of Integrative Agriculture2022年6期
Journal of Integrative Agriculture2022年6期
- Journal of Integrative Agriculture的其它文章
- A major and stable QTL for wheat spikelet number per spike validated in different genetic backgrounds
- The GhMAX2 gene regulates plant growth and fiber development in cotton
- Optimization of nitrogen fertilization improves rice quality by affecting the structure and physicochemical properties of starch at high yield levels
- Source-sink relations and responses to sink-source manipulations during grain filling in wheat
- lmage-based root phenotyping for field-grown crops:An example under maize/soybean intercropping
- Genome-wide identification,evolutionary selection,and genetic variation of DNA methylation-related genes in Brassica rapa and Brassica oleracea