
咔唑德州卟啉的合成与结构表征
2022-05-08 03:51:06雷春欣周鹏程唐艳柳谭兵斌张展吴腊梅
中南民族大学学报(自然科学版) 2022年3期
雷春欣,周鹏程,唐艳柳,谭兵斌,张展,吴腊梅
(中南民族大学 化学与材料科学学院,武汉 430074)
德州卟啉(texaphyrin)是SESSLER课题组1987年首次报道的一种扩展卟啉[1-3](如图1),是一个具有22-п电子的芳香性大环化合物,因为该环的结构与德克萨斯州的五角星旗形状类似,故而这个大环被冠名为德州卟啉[4].由于其结构与卟啉[5-6]类似,且共轭体系比18-п电子的卟啉更大,吸收波长也比卟啉更长,可达730 nm左右.而730 nm的长波能有效地穿透人体组织到达病变部位[7],非常适合病变组织的光动治疗,故而具有长波吸收的德州卟啉可作为肿瘤光动疗法的光敏剂.目前,该方面的研究已比较深入[8-9],例如lutrin就是德州卟啉络合了金属Lu3+的光敏剂药物,这类光敏剂药物对子宫癌、直肠癌都显示了良好的疗效[10-12].与卟啉相比,德州卟啉具有更大的环状空腔,可以和体积较大的金属离子如镧系金属离子高效络合,这就为德州卟啉在生物成像方面的应用提供了结构基础.事实上,德州卟啉的钆(III)配合物对于脑部肿瘤具有优良的显影效果,已在临床研究中用于脑部肿瘤的MRI(磁共振成像)造影剂[9,13].无论作为光敏剂还是造影剂,德州卟啉都显示出良好的肿瘤选择性,因为其结构与天然的卟啉(如血红素)具有非常相似的结构,因而也具有相似的生理效应.利用德州卟啉良好的肿瘤选择性,SESSLER课题组近年把德州卟啉发展为顺铂类抗癌药物的靶向载体工具,从而较大提高了毒副作用较大的顺铂类抗癌药物的选择性,在降低这类药物对正常组织的毒性的同时也提高了选择性杀死癌细胞的效率[14-15].
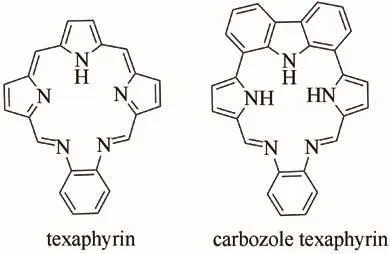
图1 德州卟啉与咔唑德州卟啉Fig.1 Texaphyrin and carbozoletexaphyrin
虽然有关德州卟啉的研究和应用已经比较成熟,但是这些研究都是基于原有的德州卟啉结构,到目前为止少有关于德州卟啉骨架结构改造或德州卟啉衍生物的研究报道.为此,本文进行了扩展德州卟啉共轭结构的尝试,设计了如图2所示的咔唑德州卟啉5的合成,即引入两个苯环单元并环在吡咯上(即咔唑单元)以扩展德州卟啉的共轭结构或改变其共轭路径,而不改变德州卟啉的希夫碱结构.具体合成及结构表征如下.
1 实验部分
1.1 样品、试剂和仪器
所有反应均在惰性气体氩气保护下进行;紫外-可见光吸收光谱采用Varian Cary 5000紫外分光光度计;核磁共振采用Bruker AV400或AV600;X-ray单晶衍射用的是Brucker D8.
1.2 中间产物及目标化合物的合成
所有化合物的合成路线及反应条件均如图2所示.
1.2.1 化合物1的合成
据文献方法合成化合物1,所得氢谱与文献一致[16].在氩气保护下,于250mL圆底烧瓶中用100 mL乙酸溶解0.81 g 3,6-二叔丁基咔唑,再往其中加入0.92 g液溴,升温至90℃反应3.5 h.反应结束后,将反应混合液稍冷却后减压旋蒸去掉大部分液体,冷却抽滤并水洗滤饼至滤液中性,干燥滤饼得到化合物1的白色固体1.20 g,产率95%.1H NMR(400 MHz,CDCl3)δ8.14(s,1H),7.98(s,2H),7.64(s,2H),1.44(s,18H).
1.2.2 化合物2的合成
在100 mL圆底烧瓶中依次加入217 mg化合物1与385.2 mg吡咯甲酸酯的硼酸酯(如图2),50 mg四(三苯基膦)钯,然后加入混合溶剂:25 mL甲苯、12 mL乙醇和2.5 mL浓度为2 mol‧L-1的K2CO3水溶液.升温至90℃后反应24 h.反应结束,分液出有机层,然后用水洗有机层.浓缩后通过柱层析法分离得到247.8 mg棕色固体化合物2,产率为75%.1H NMR(400 MHz,CDCl3)δ8.95(s,2H),8.12(s,2H),7.99(s,1H),7.46(s,2H),4.32(d,J=7.1 Hz,4H),2.83(d,J=7.4 Hz,4H),2.50(d,J=7.5 Hz,4H),1.62(s,6H),1.47(s,18H),1.35(t,J=7.1 Hz,6H),1.02(t,J=7.5 Hz,6H).13C NMR(100 MHz,CDCl3)δ161.4,143.1,136.5,134.3,130.0,125.1,124.8,123.9,118.9,116.6,115.4,60.0,34.9,32.1,18.6,17.7,16.3,15.8,14.6.

图2 咔唑德州卟啉的合成路线Fig.2 Synthesis of carbozole texaphyrin
1.2.3 化合物3的合成
665 mg化合物2与200 mg固体氢氧化钠加入到25 mL的圆底烧瓶中,加入10 mL乙二醇作溶剂.升温至180℃后反应2 h,结束反应并冷却反应液至室温然后倒入冰水中,立刻有固体析出,抽滤得468.9 mg棕色固体即化合物3,产率90%.1H NMR(400 MHz,CDCl3)δ8.05(d,J=1.6 Hz,3H),7.86(s,2H),7.44(d,J=1.6 Hz,2H),6.54(s,2H),3.73(s,1H),2.58~2.43(m,8H),1.47(s,18H),1.11(t,J=7.5 Hz,12H).13C NMR(100 MHz,CDCl3)δ142.7,136.2,129.2,128.4,126.2,124.6,123.9,122.4,115.3,34.9,32.1,18.4,18.0,16.1,14.1.
1.2.4 化合物4的合成
向100 mL圆底烧瓶中加入50 mL二氯乙烷,1 mL DMF,613.3 mg POCl3,室温下搅拌10 min.将反应体系冰浴冷却至0℃,并在冰浴下缓慢滴加由521 mg的化合物3与5 mL二氯乙烷配成的溶液,滴加完成后将反应温度升高至80℃继续反应5 h.然后冷却至0℃,缓慢加入30 mL K2CO3溶液(138.2 mg),之后再升温至80℃反应2 h.待反应结束后,将反应混合液减压蒸馏,浓缩液用二氯甲烷萃取三次,合并有机相并用水洗两次.浓缩后通过柱层析分离产物,得到536.5 mg黑色固体化合物4,产率为93%.1H NMR(600 MHz,CDCl3)δ9.69(s,2H),9.52(s,2H),8.63(s,1H),8.15(d,J=1.5 Hz,2H),7.52(d,J=1.7 Hz,2H),2.79(d,J=7.6 Hz,4H),2.58(d,J=7.5 Hz,4H),1.48(s,18H),1.28(t,J=7.6 Hz,6H),1.09(t,J=7.5 Hz,6H).13C NMR(151 MHz,CDCl3)δ142.6,136.1,128.7,125.2,124.7,124.1,116.7,114.6,34.8,31.9,17.5,15.9.
1.2.5 化合物5(咔唑德州卟啉)的合成
将115.4 mg化合物4与24.76 mg邻苯二胺加入到250 mL的烧瓶中,用100 mL甲苯与50 mL甲醇作混合溶剂,室温搅拌下缓慢滴加0.05 mL乙酸乙酯的氯化氢饱和溶液,滴加完毕后,升温至80℃,回流反应12 h.然后将反应液减压浓缩得到固体,并在甲醇与二氯甲烷体系中重结晶,得到105.1 mg淡黄色粉末状化合物5,产率为81%.1H NMR(400 MHz,CD2Cl2)δ9.48(s,1H),8.17(s,2H),8.13(d,J=1.6 Hz,2H),7.71(d,J=1.6,2H),7.25~7.19(m,4H),6.00(s,2H),2.80~2.72(m,8H),1.54(s,18H),1.32~1.26(m,12H).13C NMR(100 MHz,CD2Cl2)δ143.2,142.9,142.5,136.9,136.0,135.7,129.1,126.1,125.5,124.2,123.6,116.1,116.0,115.7,34.7,31.7,17.8,17.3,17.2,15.7.
1.3 晶体结构的检测
用甲醇、乙醇、THF、二氯甲烷、乙腈、氯仿、石油醚、正己烷等体系培养化合物5的单晶,最终在二氯甲烷-甲醇体系中通过慢速挥发法得到了无色针状化合物5的单晶.将化合物5的单晶置于X-射线单晶衍射仪上,于150(2)K,采用石墨单色化Mo Kα射线(λ=0.071 073 nm)为衍射光源,晶体结构用直接法解得,具体晶体参数见表1.
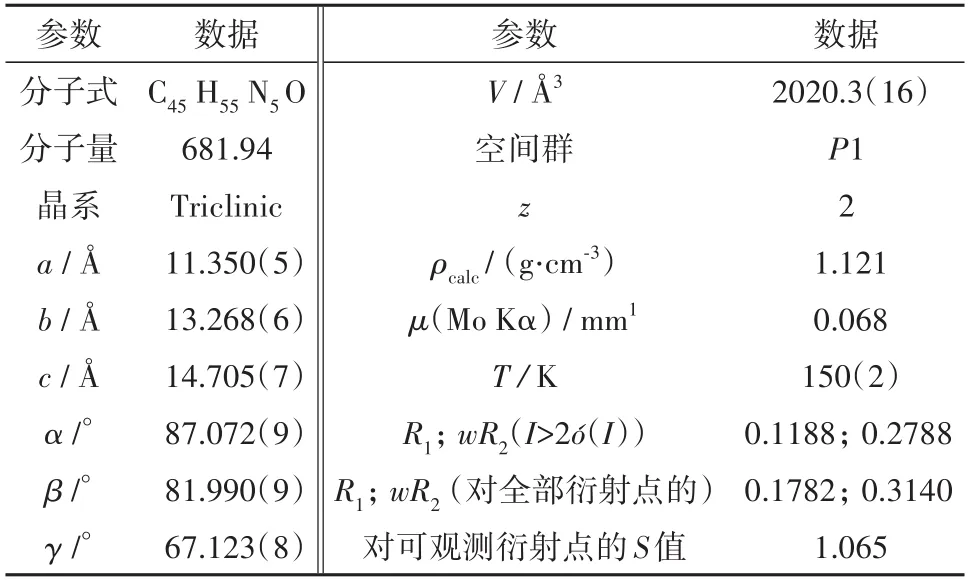
表1 化合物5的晶体参数Tab.1 Crystallographic data for compound 5
2 结果与讨论
2.1 咔唑德州卟啉的合成
以商品化的3,6-二叔丁基咔唑为原料,与N-溴代琥珀酰亚胺(NBS)或液溴在乙酸中90℃反应2 h都可生成目标化合物1,使用液溴与3,6-二叔丁基咔唑反应副产物较少、产率更高,反应的后处理也较为简单,杂质通过水洗即可除去,剩下的白色固体便是化合物1.故最终采用液溴进行溴化的反应方案.
Suzuki偶联合成化合物2的反应是整个合成路线中较为关键的一步,本文对该步反应条件进行了优化.平行实验表明:对于钯催化剂,双三苯基膦二氯化钯或二氯化钯/三苯基膦的催化效果都不够理想,只有四(三苯基膦)钯才能较好地催化该偶联反应.对于溶剂的筛选,发现以甲苯作为溶剂以乙醇作为助溶剂,反应的效果较好.在优化的催化剂和溶剂体条件下,原料化合物1基本可以反应完全,目标化合物2的产率可达到75%.在分离纯化目标产物时,通过柱层析分离得到了一种由两分子原料化合物1自耦连形成的副产物,如图3所示.
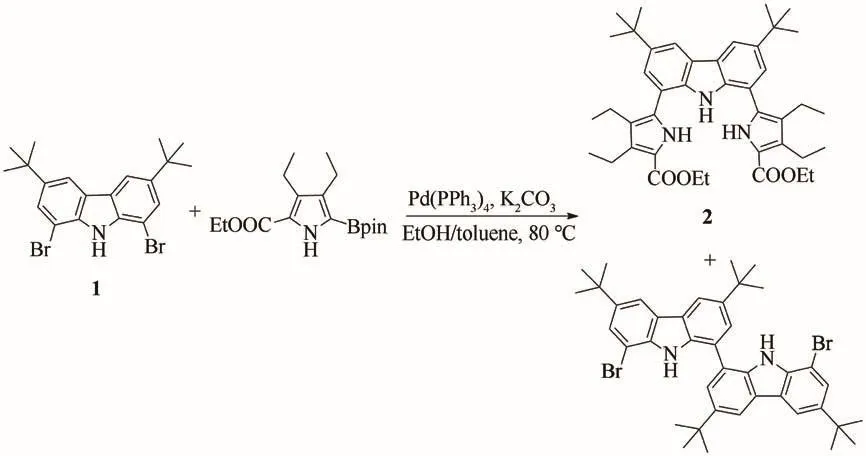
图3 化合物2及耦连产物的合成路线Fig.3 Synthesis of compound 2 and the bi-carbozole
化合物3可由化合物2在强碱高温条件下水解脱羧得到.由于化合物3的吡咯单元α位没有取代基,化学性质十分活泼,很容易被氧化.所以该脱羧反应要在惰性气体保护下进行,而且在升温至180℃后约2 h就要结束反应,否则反应时间过长会导致产物变质.化合物3不宜在溶液中保存过久,但在固体状态可以长期存放.
关键前体化合物4则可通过Vilsmeier-Haack反应由化合物3制备.进行Vilsmeier-Haack反应时,先向反应液中加入POCl3使其与DMF(N,N-二甲基甲酰胺)形成活性中间体,在此反应阶段会放出大量热量,因此需要在低温下缓慢将POCl3加入到反应液中,待DMF与POCl3反应生成活性中间体之后,再与化合物3反应,最后加入碱水解得到目标产物.由于POCl3极易发生水解,生成磷酸与盐酸,因此在反应体系中应严格进行除水并用惰性气体保护.引入醛基之后,化合物4相较于化合物3稳定性大大增强.
得到关键前体化合物4之后,本文探索了其与邻苯二胺缩合制备咔唑德州卟啉的反应.分别尝试了用HNO3、TFA、HCl、FeCl3等无机酸或路易斯酸作为催化剂,通过TLC监测反应,发现只有在使用HCl作为催化剂时,反应初期有小极性的黄色产物点出现,推测其很有可能是目标产物.然而,随着反应的进行,TLC监测到黄色点又逐渐消失,可能是由于咔唑德州卟啉是构建在可逆共价键基础上的,在酸性条件下,亚胺键很容易水解造成咔唑德州卟啉的解构.为了尽量避免新生成的咔唑德州卟啉水解解构,使用溶解在乙酸乙酯中的氯化氢代替盐酸作催化剂,最终成功合成了咔唑德州卟啉.所得粗产物可用无水二氯甲烷与甲醇进行重结晶纯化,从而得到咔唑德州卟啉5.
2.2 咔唑德州卟啉5的结构表征
2.2.1 核磁氢谱图
化合物5的核磁氢谱共有9组峰,其中化学位移为1.29左右的多重信号峰对应吡咯β位相连的多个乙基的甲基—CH3上的氢;化学位移为2.76的多重信号峰则对应于这些多个乙基的亚甲基—CH2—上的氢,出现在1.54的单峰对应咔唑环上的叔丁基上甲基的氢;7.22左右的多重信号峰对应苯环上的4个氢;而7.72与8.13处的两组峰相互远程耦合,对应咔唑环上4个氢;化学位移为8.17的单峰对应两个亚胺键的2个氢;6.00处的小包峰为活泼氢的信号峰,它们对应两个吡咯上的的2个氮氢;9.48出现的信号峰,为咔唑环氮上的一个氢.大环孔穴内的氮氢信号峰没有出现在高场屏蔽区,表明该化合物没有形成芳香性共轭体系.
由化合物5的核磁碳谱和其碳氢直接相关谱HSQC谱图,可知碳谱中34.7的碳在HSQC中无氢对应,应该为叔丁基的叔碳.15.7的碳与1.29左右的多重峰的氢直接相连,所以为乙基中甲基碳,依次类推17.3的碳是乙基中—CH2—上的碳,31.7的碳与单峰的18个氢相连,是叔丁基中的甲基碳,115.6的碳与8.13的氢相连,123.6的碳与7.72的氢相连,143.2的碳与8.17的氢相连,116和125左右的碳与7.22左右的氢相连.另外从杂核多键氢碳相关谱HMBC可以发现8.13的氢分别与34.7,123.6,136.0的碳相关,7.72的氢分别与34.7,115.6,135.7的碳相关.说明8.13的氢是咔唑环4,5位碳上的氢而7.72的氢是咔唑环上的2,7位上的氢,7.22左右的氢为苯环上的4个氢.
2.2.2 紫外-可见吸收
如图4所示,咔唑德州卟啉5的紫外可见最大吸收波长在336 nm处,体现出典型的非共轭大环化合物的光学吸收特征,既没有明显的S吸收带,也没有出现吸收波长大于600 nm的Q带吸收峰.该化合物呈淡黄色,而非卟啉及类似物的深紫色或深绿色,这些现象都支持该化合物没有形成有效的共轭芳香体系.

图4 咔唑德州卟啉的紫外-可见吸收光谱Fig.4 UV-vis of carbozole texaphyrin
2.2.3 X-射线晶体数据
咔唑德州卟啉5的晶体结构如图5(a)所示.显而易见,化合物5分子具有非平面的、类似马鞍的结构,其中的两个吡咯单元平面都和咔唑单元平面具有一定的夹角(其二面角分别为:42.67°与41.18°).而来自邻苯二胺的苯环既不和吡咯单元也不和咔唑单元共平面.根据休克尔规则的共平面原则,可以判定化合物5不具有芳香性结构,这和该化合物的紫外与核磁的数据提供的信息相一致.
值得注意的是在化合物5的单晶结构中(注:在二氯甲烷-甲醇中通过慢速挥发法生长得到),每个咔唑德州卟啉分子还结合了一分子的甲醇分子.根据单晶结构可以清晰判断出该甲醇分子与两个吡咯单元的NH基团及两个亚胺氮形成了有效的氢键(O1…N2距离为2.936Å;O1…N5距离为2.970Å;N3…O1距离为2.919Å;N4…O1距离为2.912Å).此外,咔唑单元的NH基团与相邻但翘起的吡咯单元的氮原子也形成了稍弱的分子内氢键(N2…N1距离为3.075Å;N5…N1距离为3.036Å).详细氢键如表2及图5(b)所示.这些固体状态下存在的氢键特别是甲醇分子与咔唑德州卟啉的分子间氢键表明德州卟啉有可能是良好的阴离子络合主体分子,甚至有可能可以用于调节细胞内某一阴离子的浓度,这方面的研究目前正在进行之中.
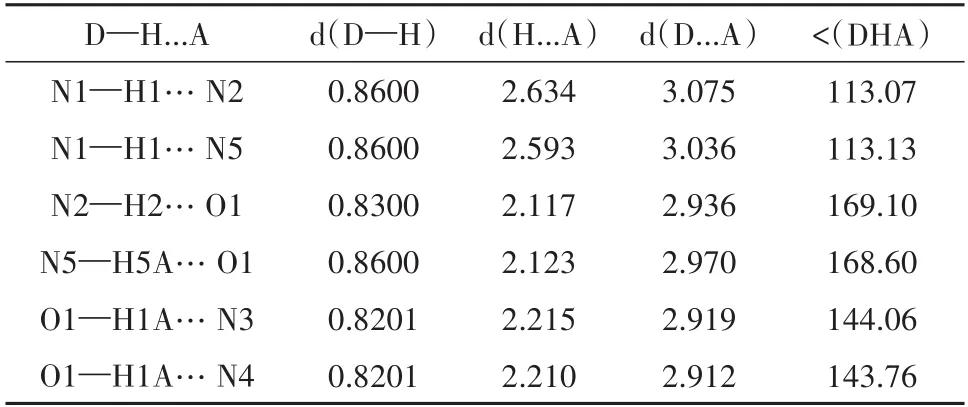
表2 化合物5的氢键键长(Å)及键角(°)Tab.2 Hydrogen bond length(Å)and bond angle(°)for compound 5

图5 化合物5的单晶结构Fig.5 Single crystal structure of compound 5
3 结语
本文报道了由3,6-二叔丁基咔唑为原料,经过五步反应合成咔唑德州卟啉的方法,并通过1H NMR、13C NMR、紫外-可见及单晶X-衍射方法对咔唑德州卟啉进行了结构表征,为卟啉类化合物的合成与研究提供了一些有效的方案.虽然目前产物没能形成环状共平面的芳香性结构,没有得到具有近红外吸收性能的类卟啉化合物,但该研究为类卟啉化合物的阴离子络合开辟了研究空间.
猜你喜欢
辽宁化工(2022年8期)2022-08-27 06:03:04
云南化工(2021年5期)2021-12-21 07:41:12
石油炼制与化工(2021年12期)2021-12-14 06:26:38
四川轻化工大学学报(自然科学版)(2021年3期)2021-08-30 06:36:44
塑料助剂(2018年6期)2018-03-25 05:59:16
山东化工(2018年1期)2018-03-10 02:56:49
合成化学(2015年10期)2016-01-17 08:56:06
应用化工(2014年3期)2014-08-16 13:23:50
华东师范大学学报(自然科学版)(2014年4期)2014-03-11 16:18:28
无机化学学报(2014年4期)2014-02-28 17:31:16
