
Biomarkers for therapy selection in metastatic urothelial cancer
2022-05-05 08:54:34TomiJunJonathanAnkerMatthewGalsky
Tomi Jun, Jonathan Anker, Matthew D.Galsky
1Division of Hematology and Medical Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
2Medical Sciences, Sema4, Stamford, CT 06902, USA.
3Division of Hematology and Medical Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, USA.
Abstract The treatment of metastatic urothelial cancer (mUC) has been transformed by recent progress in clinical trials and drug development.There are now three therapeutic classes with proven benefits in mUC: chemotherapy,immunotherapy, and targeted therapy.The optimal sequence and combination of these classes remain to be defined.Biomarker development is essential to guide treatment selection at each therapeutic juncture.Two biomarkers, programmed death-ligand 1 expression and fibroblast growth factor receptor alterations, have been incorporated into the mUC treatment paradigm thus far.This review discusses predictive biomarkers in development and their potential to influence mUC treatment selection moving forward.
Keywords: Metastatic urothelial cancer, biomarkers, immunotherapy, chemotherapy, targeted therapy, antibodydrug conjugates
INTRODUCTION
For 30 years, cisplatin-based combination chemotherapy was the only treatment to demonstrate a meaningful overall survival (OS) benefit in metastatic urothelial cancer (mUC)[1].In subsequent lines,patients were treated with single-agent chemotherapy with limited benefit[1].This sparse treatment landscape has been transformed by contemporary trials demonstrating OS benefits from anti-PD-1/programmed death-ligand 1 (PD-L1) immune checkpoint inhibitors (ICIs) and the antibody-drug conjugate (ADC) enfortumab vedotin (EV)[2-4].Promising early-phase results have also led the United States Food and Drug Administration (FDA) to grant accelerated approval for sacituzumab govitecan (SG)[5,6],another ADC, and the tyrosine kinase inhibitor (TKI) targeting fibroblast growth factor receptor(FGFR)1-4, erdafitinib[7].
The expansion of mUC treatment options raises the question of treatment selection.How can therapeutic options be prioritized to maximize benefits and minimize harms for individual patients? One answer to this question is: biomarkers.Ideally, biomarkers guide clinical management by clearly distinguishing patients who will or will not benefit from treatment.This review will discuss molecular biomarkers in clinical use and in development for treatment selection in metastatic urothelial cancer.
BIOMARKER USES AND DEVELOPMENT
While a comprehensive description of biomarker types and their development is outside the scope of this review, a brief overview may be useful to situate the various biomarkers described herein along the axis of predictivevs.prognostic and along the continuum from exploratory to clinically validated.
A biomarker can be predictive, prognostic, or both.Predictive biomarkers are associated with outcomes in the context of specific treatments, while prognostic biomarkers are associated with outcomes irrespective of treatment.This treatment-dependent distinction implies that a biomarker’s classification may change as a disease’s treatment landscape evolves.This is exemplified by the transformation of human epidermal growth factor receptor 2 (HER2) amplification in breast cancer from an adverse prognostic marker into a treatment-defining predictive marker with the advent of effective anti-HER2 agents.Today’s prognostic markers may be tomorrow’s drug targets.
As this review is concerned with biomarkers that can guide treatment selection, our focus is on predictive biomarkers.Aside from two predictive biomarkers routinely used in mUC management - PD-L1 andFGFRalterations - the other candidate biomarkers discussed here have yet to be prospectively validated for treatment selection.These candidate biomarkers typically have demonstrated associations with outcomes in retrospective cohorts or post-hoc analyses of clinical trials and may havein vitrodata to support a mechanistic link between the biomarker and treatment outcomes.Where possible, we will highlight biomarkers being incorporated into prospective trials, as these have the greatest potential to become clinically validated.
To formally establish a biomarker as predictive, there should be a comparison of outcomes by both treatment arm (experimentalvs.control) and biomarker status (positive or negative).Without a control arm, it is not possible to determine whether the biomarker’s association with outcomes is specific to the experimental treatment (predictive) or a general phenomenon (prognostic).That said, in some cases, the mechanistic link between the predictive biomarker and the treatment’s mechanism is seen as strong enough to justify running a prospective trial in a biomarker-selected population, effectively assuming that there would be no benefit from the treatment in a biomarker-negative population.
The treatment landscape, the mechanistic link between biomarker and treatment, and the level of evidence available should all be considered when evaluating the clinical utility of a novel biomarker.
CHEMOTHERAPY BIOMARKERS
Cisplatin-based combination chemotherapy regimens are the backbone of first-line mUC treatment.Randomized clinical trials published in the 1990s demonstrated that the combination of methotrexate,vinblastine, doxorubicin, and cisplatin (MVAC) improved OS compared to cisplatin alone and compared to the combination of cisplatin, cyclophosphamide, and doxorubicin[8,9].Subsequently, the combination of gemcitabine and cisplatin (GC) was shown to have similar survival outcomes as MVAC, with a more favorable safety profile[10].
At present, despite the introduction of novel agents for mUC, cisplatin-based chemotherapy remains the standard front-line treatment for advanced UC in patients without contraindications to it [Figure 1].Immunotherapy has a role as maintenance treatment after chemotherapy[2], but upfront chemoimmunotherapy combinations and immunotherapy-only combinations have not shown OS benefits compared to chemotherapy alone[11-13].The chemotherapy-free regimen of enfortumab vedotin plus pembrolizumab (EV/P) has shown promising early-phase results in the front-line cisplatin-ineligible setting[14]and is being tested against upfront chemotherapy in an unselected mUC population in the EV-302 phase III trial (NCT04223856).In this evolving treatment landscape, chemotherapy biomarkers would be valuable to distinguish responders - likely well-served by the current paradigm of upfront chemotherapy -from non-responders, who may benefit from novel combinations or alternative agents.
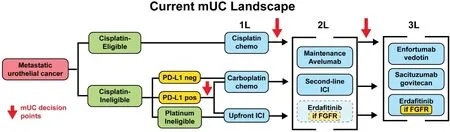
Figure 1.Current landscape.Current metastatic urothelial carcinoma (mUC) treatment landscape.Clinical factors which influence treatment selection, such as cisplatin-eligibility, are highlighted in green.Biomarkers are highlighted in yellow.Treatment options are shown in blue.In the second line, erdafitinib is an FDA-approved option but results from the phase III THOR trial (NCT03390504)comparing it against pembrolizumab are not yet available, thus it is shown with a dotted outline.PD-L1: Programmed death-ligand 1; ICI:immune checkpoint inhibitor; 1L: first-line; 2L: second-line; 3L: third-line.
It is worth noting that most of the data concerning chemotherapy biomarkers discussed below come from studies of muscle-invasive bladder cancer (MIBC).Neoadjuvant chemotherapy (NAC) regimens for MIBC are similar to regimens for mUC.However, until recently, a chemotherapy response biomarker was arguably more useful in the context of MIBC since the complete response to NAC has prognostic implications which may guide further treatment.
DNA damage repair pathways
Cisplatin binds to DNA, creating adducts and crosslinks that inhibit transcription and replication, and may lead to apoptosis.In normal cells, multiple DNA damage repair (DDR) pathways are activated in response to these lesions and cooperate to repair them[15].These include nucleotide excision repair (NER),homologous recombination repair (HRR), and Fanconi anemia (FA) pathways.Mutations in each of these pathways, or in genes regulating these pathways, have been linked to platinum sensitivity in bladder cancer.
Nucleotide excision repair
The NER pathway recognizes bulky DNA adducts, such as those formed by cisplatin and ultraviolet radiation, and excises them[16].Germline mutations in NER genes cause xeroderma pigmentosum, an autosomal recessive disorder characterized by marked photosensitivity.In fact, several NER genes (XPA-G)were initially named for their association with xeroderma pigmentosum.Just as germline mutations in NER genes can predispose individuals to UV sensitivity, tumor somatic mutations in NER pathway genes such as excision repair cross-complementation group 1 and 2 (ERCC1andERCC2) can also alter tumor cisplatin sensitivity.
ERCC1 expression levels
ERCC1is a key NER gene linked to cisplatin sensitivity in a variety of solid tumors.The ERCC1 protein heterodimerizes with ERCC4 to form an endonuclease complex which participates in the excision of the damaged DNA.Lower ERCC1 levels, measured by mRNA expression or immunohistochemistry (IHC),have been correlated with cisplatin sensitivity in lung, cervical, ovarian, gastric, and colorectal cancers[17-22].While most of these observations were made in small or retrospective cohorts, the IALT Bio study involved a cohort of 761 lung cancer patients who had participated in a prospective study of adjuvant cisplatin-basedchemotherapyvs.observation.In this cohort, the authors found that the absence of ERCC1 staining by IHC was predictive of a benefit from adjuvant chemotherapy (test for interaction,P= 0.009)[19].
In urothelial carcinoma, low ERCC1 levels by either mRNA quantification or IHC have also been associated with improved response to cisplatin in the perioperative and metastatic settings[23-34].In 2007,Bellmuntet al.[34]reported a retrospective analysis of 57 mUC patients treated with GC-based regimens; low levels ofERCC1mRNA expression correlated with improved OS (25.4 monthsvs.15.4 months,P= 0.03).Subsequently, a 2017 meta-analysis of 13 studies concluded that ERCC1 positivity was associated with worse progression-free survival (PFS; pooled HR = 1.54, 95%CI: 1.13-211,P= 0.006), with a non-significant trend towards worse OS (pooled HR = 1.63, 95%CI: 0.93-2.88,P= 0.09)[35].However, this meta-analysis was limited by a lack of relevant prospective studies and variation in the methods of ERCC1 detection.
ERCC2 mutations
ERCC2is mutated in about 10% of muscle-invasive urothelial carcinomas[36].It encodes a helicase subunit of the Transcription factor II Human complex, which plays a key role in the NER pathway by unwinding DNA at the site of damage prior to excision of the damaged bases.The majority ofERCC2mutations in UC affect the helicase domain andin vitrodata suggest that these mutations impair NER activity and confer cisplatin sensitivity[37,38].
Mutations inERCC2have been associated with complete pathological responses and improved OS after neoadjuvant cisplatin-based chemotherapy in two independent MIBC cohorts[38,39].Similar, though nonsignificant, trends have been observed in other neoadjuvant MIBC cohorts[40-42].In the metastatic setting, a recent study including a mix of patients with locally advanced (n= 62) and metastatic UC (n= 245)reported that theERCC2-associated mutation signature SBS5, but notERCC2mutations, was correlated with response[43,44].
In the context of chemo-immunotherapy, the ongoing HCRN GU 16-257 study recently reported thatERCC2mutations were associated with clinical and pathological complete response in MIBC patients treated with neoadjuvant gemcitabine, cisplatin, and nivolumab[45].
Homologous recombination
HRR is a high-fidelity DNA repair mechanism, which is involved in the repair of double-stranded breaks and interstrand crosslinks.In HRR, the undamaged homologous chromosome serves as a template for the repair of the damaged strand.BRCA1andBRCA2are perhaps the most widely recognized HRR genes,known for their roles as cancer predisposition genes and as predictive biomarkers for sensitivity to poly ADP-ribose polymerase (PARP) inhibitors and platinum-based chemotherapy.
The predictive association betweenBRCA1/2mutations and platinum chemotherapy is best established in breast and ovarian cancer[46-48].In breast cancer, the phase III TNT trial found a significant interaction betweenBRCA1/2mutation status (either germline or somatic) and response to carboplatin; the objective response rate for carboplatin amongBRCA-mutated tumors was twice that ofBRCA-wild-type tumors (68%vs.33%,P= 0.03)[48].In ovarian cancer, the mechanistic link betweenBRCA2deficiency and platinum sensitivity is illustrated by the observation that mutations which restore wild-typeBRCA2function also confer platinum resistance[47].
BRCAmutations can also be found in urothelial cancer and may be associated with platinum sensitivity.SomaticBRCA1/2alterations were present in 19% of MIBC samples in TCGA (The Cancer Genome Atlas),and germlineBRCA1/2variants have been observed in 2%-4% of UC patients[36,49,50].In a recent multi-omic analysis of 300 patients with MIBC or mUC,BRCA2mutations were associated with the single base substitution 5 (SBS5) mutation signature and with chemotherapy response[43].In contrast toBRCA2mutations,BRCA1/2expression levels have not been associated with outcomes in retrospective platinumtreated UC cohorts[34,51].
RAD51(RAD51 recombinase) is another HRR gene, which is being investigated as a chemotherapy biomarker in UC.In the context of double-stranded breaks, the RAD51 protein (in conjunction with BRCA2) coordinates the process matching the damaged strands to their homologous complements[52].High nuclear staining for RAD51 has been associated with worse OS for mUC patients treated with cisplatinbased chemotherapy[51].
Other DDR genes
OtherDDRgenes have been linked to cisplatin sensitivity.A 2015 study by Plimacket al.[53]used decision tree analysis to identify tumor mutations that could efficiently classify MIBC patients by cisplatin responsiveness.Out of 287 cancer-related genes on the sequencing panel, their analysis found that mutations inATM(ATM serine/threonine kinase),RB1(RB transcriptional corepressor 1), orFANCC(FA complementation group C) most clearly separated pathologic responders from non-responders in two clinical trial cohorts.An updated report showed that 5-year OS was 85% in those withATM/RB1/FANCCmutations compared to 46% in those without[54].ATMandRB1are cell cycle regulators in response to DNA damage, whileFANCCis a member of the core FA complex that is critical to interstrand crosslink repair.
Mutations in DDR genes as a class have also been associated with platinum response.In a 2017 retrospective analysis, Teoet al.[55]found that alterations in a panel of 34 DDR genes (representing various pathways)were associated with improved PFS and OS in platinum-treated mUC patients.The most commonly mutated DDR genes wereATM(n= 18),BRCA2(n= 13),BRCA1(n= 9), andNBN(n= 10), a component of the MRN complex that recognizes double-stranded breaks and initiates HRR.
Other somatic alterations
Groenendijket al.[41]foundERBB2(erb-b2 receptor tyrosine kinase 2) missense mutations (but notERBB2amplification) to be associated with response to platinum-based NAC in a retrospective study of 38responding and 33 non-responding MIBC patients.There were 9ERBB2mutations among the responders and none among the non-responders (24%vs.0%,P= 0.003).Van Allenet al.[38]had only 3ERBB2-altered samples in their cohort, but all 3 were responders (P= 0.12).Conversely, Plimacket al.[53]did not findERBB2alterations to be associated with response, though they did not report separate analyses forERBB2missense mutationsvs.amplification.Further analysis is required to determine whetherERBB2missense mutations are a potential marker of chemosensitivity.
Molecular classifications
The extraction and quantification of tumor RNA enable tumors to be described by their gene expression profiles.This opens another avenue to differentiate tumors at the molecular level and potentially identify subgroups with differential response to antineoplastic agents.Clustering algorithms make it possible to group tumors into molecular subtypes based on similarities in gene expression.
Several efforts have been made to establish molecular taxonomies of UC, mostly based on nonmetastatic or MIBC samples[36,56-70].However, the molecular subtypes generated by different groups can vary based on differences in cohorts, algorithms, and analytic decisions made by the investigators.Recently,Kamounet al.[71]reported a collaborative effort to integrate previously published classification schemes resulting in six consensus molecular subtypes for MIBC: basal/squamous (35%), luminal papillary (24%),luminal unstable (15%), luminal nonspecified (8%), stroma-rich (15%), and neuroendocrine-like (3%).
The distinction between luminal and basal UC is a recurring theme across molecular subtyping studies.These subtypes were initially named after their similarity to basal and luminal breast cancer subtypes[64,65,70].Basal UC is associated with squamous differentiation and expression of basal cytokeratins such asKRT5,KRT6, andKRT14.The luminal subtype is characterized by the expression ofKRT20 (keratin 20),GATA3(GATA binding protein 3), andFOXA1(forkhead box A1) and is associated withFGFR3alterations and papillary histology.The consensus subtyping further divides luminal into three subtypes: luminal papillary,associated with a non-invasive papillary signature andFGFR3transcriptional activity; luminal nonspecified,associated with increased fibroblastic infiltration; and luminal unstable, associated with genomic instability and increased cell cycle activity[71].
The utility of molecular subtypes as predictive biomarkers of chemotherapy response is unclear, and studies have produced conflicting results.Choiet al.[64]previously described a p53-like subtype that was chemoresistant in small retrospective cohorts.This subtype was associated with the worst outcomes in a singlearm trial of neoadjuvant MVAC with bevacizumab[72].The p53-like subtype falls under the stroma-rich consensus subtype, but none of the consensus subtypes were found to associate with NAC response in the Kamounet al.[71]publication.
Some studies have suggested that basal UC is a chemo-sensitive subtype[64,72,73].Seileret al.[73]observed that patients with basal-type tumors had the greatest benefit from NAC compared to other subtypes, though they did not formally test for an interaction between NAC and subtype.The basal subtype was also associated with the best OS in the trial of neoadjuvant MVAC with bevacizumab mentioned above[72].On the other hand, Taberet al.[43]recently reported that the basal/squamous consensus subtype was associated with reduced NAC response in a cohort of 300 advanced UC patients.
Another approach to developing gene expression-based predictive biomarkers, known as coexpression extrapolation (COXEN), has been proposed by Smithet al.[74].The COXEN approach identifies gene expression signatures in cancer cell lines associated within vitrochemotherapy sensitivity and extrapolatesthose signatures to predict chemosensitivityin vivo.COXEN gene expression models for response to cisplatin-based NAC regimens were tested in the phase II SWOG S1314 trial, but failed to predict response to either GC or ddMVAC[75].
Clinical trials examining chemotherapy biomarkers
The CALGB 90601 study (NCT00942331) was a phase III trial that enrolled 506 patients and compared gemcitabine and cisplatin with or without bevacizumab for the front-line treatment of mUC.There was no significant difference in the primary outcome of OS between the two arms[76].However, the trial protocol also included correlative sub-studies aimed at testing potential chemotherapy biomarkers, including tumor expression of ERCC1, RAD51, RRM1 (ribonucleotide reductase catalytic subunit M1), BRCA1, BRCA2, and caveolin-1; somatic mutations inERCC2; and the MD Anderson molecular subtypes (basal, luminal, p53-like)[77].These results have yet to be reported.
Two ongoing phase II studies are testing biomarker-guided bladder-preservation strategies in MIBC treated with NAC (NCT03609216, NCT02710734).In these trials, patients with DDR mutations (e.g.,ATM,RB1,FANCC,ERCC2) with complete clinical responses after neoadjuvant cisplatin-based chemotherapy can be actively monitored instead of undergoing cystectomy.An interim analysis of the RETAIN (Risk Enabled Therapy After Initiating Neoadjuvant Chemotherapy for Bladder Cancer) study reported that 33 of 71 patients had a DDR mutation of interest, with 28 opting for active surveillance[78].In the HCRN GU 16-257 study, MIBC patients were treated with neoadjuvant gemcitabine, cisplatin, and nivolumab;ERCC2mutations were associated with clinical and pathological complete response[45].While these studies are in MIBC rather than mUC, positive data supporting a role for DDR genes as biomarkers in this setting could motivate efforts to define their role in treatment selection for mUC.
IMMUNOTHERAPY BIOMARKERS
ICIs have had a major impact on the treatment of advanced UC.Between 2016 and 2017, several early-phase clinical trials reported response rates of approximately 20% for single-agent anti-PD-1/PD-L1 in mUC[3,79-84].Many randomized trials have followed, seeking to define the optimal role for single-agent ICIs and to test ICIs in combination with chemotherapy, targeted therapies, or other ICIs targeting different immune checkpoints.
At present, the strongest evidence for ICI benefit in mUC is in the post-platinum chemotherapy setting.Two randomized trials - KEYNOTE-045 and JAVELIN Bladder 100 - have demonstrated OS benefits for single-agent ICIs as either second-line or maintenance therapy after platinum chemotherapy[2,3].Both trials met their primary OS endpoints in biomarker-unselected, all-comer populations.Notably, second-line ICI therapy was permitted in the control arm of JAVELIN Bladder 100 and was received by approximately onethird of control patients[85].Consequently, in the post-platinum setting, maintenance ICI rather than ICI at progression is considered the preferred approach in NCCN (National Comprehensive Cancer Network)guidelines, though there may be a risk of overtreating some patients with this approach.
Exploratory analyses have found associations between higher tumor mutation burden (TMB) and PD-L1 expression with improved OS benefit from maintenance avelumab[86].Gene expression signatures reflecting the state of the tumor immune microenvironment may also help stratify patients, though the clinical availability of such assays is limited at present[86].Separately, the concept of minimal residual disease -measured via circulating tumor DNA (ctDNA) - has shown promise in selecting patients with MIBC for adjuvant ICI[87,88].A ctDNA-based approach could be explored as a means of risk-stratifying mUC patients after platinum-based chemotherapy and selecting higher-risk patients for maintenance ICI.
Another consideration in the post-chemotherapy setting is when erdafitinib should be incorporated for patients with qualifyingFGFR2/3alterations and whether it should be prioritized over ICIs.FGFRalterations may be a negative ICI biomarker since they are associated with the luminal-papillary molecular subtype, which exhibits lower PD-L1 expression and decreased immune infiltration[71].On the other hand,immunosuppressive stromal signaling appears to be reduced inFGFR3-altered tumors, and similar ICI response rates have been observed regardless ofFGFR3status in three separate cohorts[89,90].Interestingly, in the biomarker-driven BISCAY study, the combination of durvalumab with the FGFR inhibitor AZD-4547 yielded similar response rates and outcomes as AZD-4547 alone (28.6%vs.31.3%), suggesting a limited additional benefit from durvalumab in anFGFR-altered population[91].The THOR phase III trial(NCT03390504) will definitively compare erdafitinib against pembrolizumab as a second-line treatment in patients withFGFRalterations.
In the front-line setting, ICI monotherapy has demonstrated activity in cisplatin-ineligible patients in earlyphase trials[80,82].However, the role of ICIs relative to carboplatin chemotherapy in this population is still evolving.For patients who are not candidates for any chemotherapy, ICI monotherapy is an appealing option.However, for those who can tolerate carboplatin chemotherapy, a maintenance ICI approach (per JAVELIN Bladder 100) may be favored over upfront ICI monotherapy, given its proven OS benefit.
PD-L1 expression is used as a biomarker among cisplatin-ineligible patients to guide the choice of upfront ICI monotherapyvs.carboplatin chemotherapy.Cisplatin-ineligible patients with PD-L1-low tumors should not be treated with upfront ICI monotherapy due to evidence of increased early mortality among PD-L1-low patients treated with upfront ICI compared to chemotherapy in the IMvigor130 and KEYNOTE-361 trials[11,12,92].Cisplatin-ineligible patients with PD-L1-positive tumors have the option of upfront ICI or chemotherapy followed by maintenance ICI; these options have not been directly compared in clinical trials.There may be a role for other biomarkers of chemotherapy response (e.g., DDR mutations)or ICI response (discussed below) to inform the choice of upfront ICI or chemotherapy in this setting.However, as it stands, PD-L1 expression is the only ICI biomarker that has been incorporated into mUC regulatory approvals and treatment guidelines.
ICI biomarker development has lagged in therapeutic innovation driven by clinical trials.In the absence of reliable and selective ICI biomarkers, clinical trials are testing ICI-based combinations in biomarkerunselected populations.Results to date have been mixed for the combination of chemotherapy with ICI(chemo-ICI) and negative for dual ICI-ICI therapy.Pembrolizumab with platinum-based chemotherapy did not improve PFS or OS over chemotherapy alone in KEYNOTE-361[12].Conversely, PFS was improved by combining atezolizumab with platinum-based chemotherapy in IMvigor130; OS results are still pending[11].The ICI-ICI combination of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) failed to improve OS compared to platinum chemotherapy in the DANUBE trial[13].
Moving forward, we can expect results from trials testing ICI-ICI combinations with or without chemotherapy and trials testing chemotherapy-free combinations of ICIs and targeted therapies [Figure 2].Examples of the former include the NILE (NCT03682068) and CheckMate 901 (NCT03036098) trials.NILE is comparing durvalumab plus chemotherapy, durvalumab plus tremelimumab plus chemotherapy, and chemotherapy alone in the upfront mUC setting.Due to the negative results in a biomarker-unselected population in DANUBE, the primary endpoint of NILE has been revised to focus on patients with PD-L1-expressing tumors[93].As for chemotherapy-free regimens, the most promising at present is the combination of EV/P, which has produced overall response rates of 73.3% in the first-line cisplatin-ineligible setting and has been granted FDA breakthrough designation[14].The combination will be tested against upfrontchemotherapy in phase III EV-302 trial (NCT04223856) and is being tested in additional settings in the multi-cohort EV-103 study (NCT03288545).
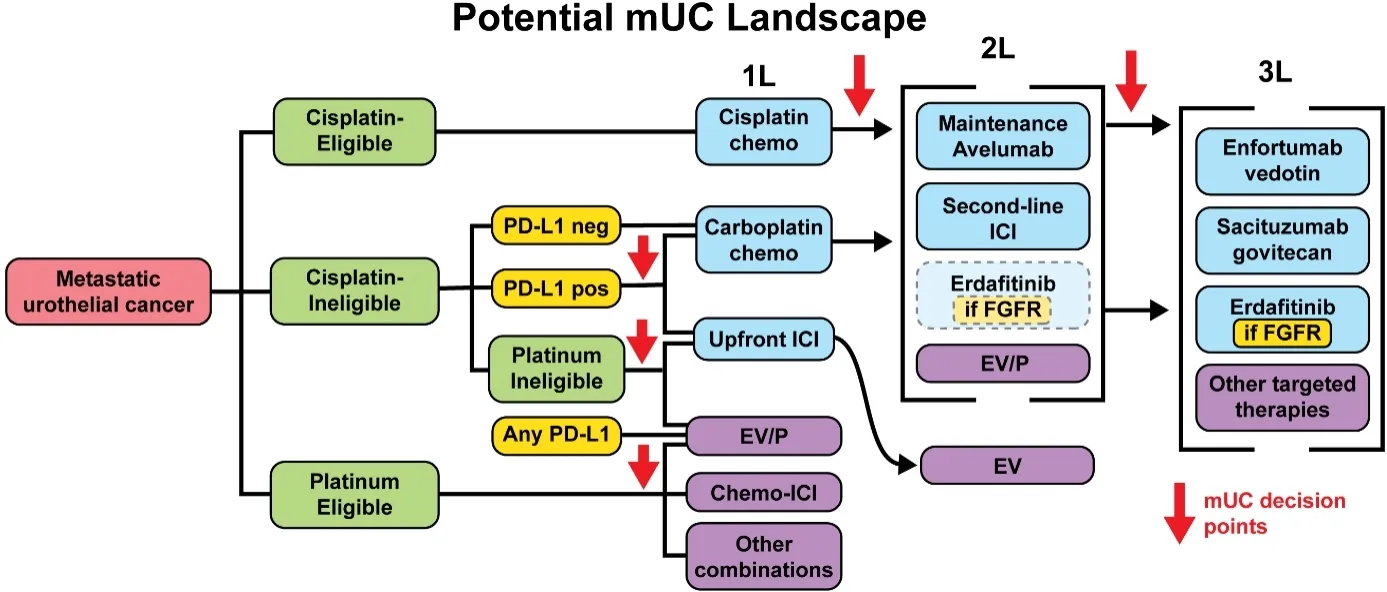
Figure 2.Potential landscape.Potential metastatic urothelial carcinoma (mUC) treatment landscape.Clinical factors which influence treatment selection, such as cisplatin-eligibility, are highlighted in green.Biomarkers are highlighted in yellow.Current FDA-approved treatments are in blue while investigational treatment options are in purple.The combination of chemotherapy and atezolizumab(Chemo-ICI) showed PFS benefits in the IMvigor130 (NCT02807636) trial but OS results are still pending.The combination of enfortumab vedotin with pembrolizumab (EV/P) is being tested in various settings, including first-line against platinum chemotherapy(EV-302, NCT04223856), first-line cisplatin-ineligible (EV-103, NCT03288545), and second-line (EV-103; EV-201, NCT03219333).Other combinations and targeted therapies are under investigation.PD-L1: Programmed death-ligand 1; ICI: immune checkpoint inhibitor; 1L: first-line; 2L: second-line; 3L: third-line.
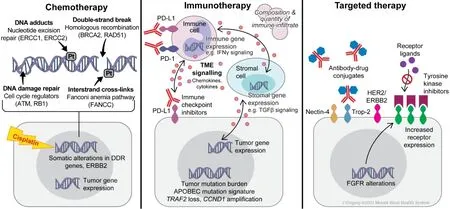
Figure 3.Treatment Classes.Summary of biomarkers for metastatic urothelial carcinoma by therapeutic classes.
While synergistic combinations are possible, it has been argued that drugs in combination regimens often act independently[85].That is, the benefit any given patient derives from a combination of A and B is due entirely to either A or B.On a population level, combination regimens improve outcomes by pooling together A-responders and B-responders.In this model, the combination of A and B is effectively a workaround for the lack of a biomarker, for if a biomarker were available, A-responders and B-responders would be treated with their most effective single agent.In practice, there may be other benefits from independently acting combinations, e.g., patients with progression may deteriorate clinically and become unable to tolerate an effective next line of therapy.Nevertheless, there is a clear need for better ICI biomarkers to guide the integration of ICIs into each patient’s treatment course.
PD-L1
Meta-analyses of prospective trials show that, overall, PD-L1 expression is associated with radiographic response to ICIs in mUC patients[94,95].However, even among PD-L1 positive patients, single-agent ICI response rates are low and variable across randomized trials, ranging from 20%-40%[3,11-13,96].
Several technical and biological factors may limit the performance of PD-L1 expression as a biomarker.PDL1 expression is measured through IHC staining.However, IHC assays differ in both the antibody used and in how the resulting stains are scored.Scoring systems differ on whether PD-L1 staining is scored on tumor cells, immune cells, or both, and on the threshold value for PD-L1 positivity.Furthermore, nearly every anti-PD-1/PD-L1 agent has been co-developed with its own assay, making it difficult to compare “PD-L1 positive” subgroups across trials.Efforts to compare PD-L1 assays in non-small cell lung cancer have found consistent staining across some assays, but not others[97,98], and have described limited inter-rater reliabilitywhen scoring PD-L1 staining on immune cells[99].
In addition to discrepancies between PD-L1 assays, there may also be heterogeneity of PD-L1 expression within tumors and between primary and metastatic sites, which is not captured by limited biopsies.Clonal evolution of tumors over time and over the course of treatment is also impractical to monitor using invasive biopsy procedures.Emerging non-invasive technologies, such as liquid biopsy and immune-targeting tracers for positron emission tomography (ImmunoPET)[100-102], may circumvent some of these limitations and pave the way for more comprehensive and longitudinal monitoring of PD-L1 or other ICI biomarkers.
Mutation burden
Bladder cancers are among the most highly mutated cancers[103].Nonsynonymous tumor mutations can lead to the formation of novel peptide sequences, or neoantigens, which the immune system recognizes as foreign and serve as the targets for immune-mediated tumor cell killing.TMB has been linked to ICI response across cancer types and in mUC specifically[79,80,104-106].
In 2020, pembrolizumab received accelerated approval from the FDA for any solid tumor with TMB ≥ 10 mutations/Mb without satisfactory treatment alternatives[107].This indication is less relevant to mUC since ICIs are already routinely integrated into the treatment plan.However, TMB may be useful as an adjunct to other ICI biomarkers.Notably, TMB is not correlated with PD-L1 expression[108].Exploratory analyses of prospective trials in mUC suggest that the combination of TMB and PD-L1 may more effectively distinguish ICI responders and non-responders than either biomarker alone[109,110].
Challenges in implementing TMB as a biomarker include selecting an optimal cutoff and harmonizing assays.Although the pembrolizumab approval used a uniform cutoff of 10 mutations/Mb across all tumors,tumor-specific cutoffs may be more appropriate[104,111].Efforts have also been made to harmonize TMB measurements, which may vary depending on the size and composition of the panel used for sequencing[112].
In addition to the quantity of mutations, there may also be qualitative aspects of mutations that contribute to immunogenicity.For example, mutation clonality may matter - whether the mutation is present in all cancer cells (clonal) or only a subset of cells (subclonal)[95].Short insertion-deletion (indel) mutations may be more likely to generate frameshift mutations which create a greater quantity of immunogenic neoantigens[113].Neoantigens and their interaction with the patient’s HLA alleles may also play a role[95,105,114].Mutational signatures, which reflect underlying mutagenic processes at work in tumors, have also been linked to ICI response.Mutational signatures attributed to the APOBEC family of cytidine deaminases are common in bladder cancers[36,103]and have been linked with favorable ICI response in prospective mUC trials[95,105,110].
According to a recent meta-analysis by Litchfieldet al.[95], which combined data from over 1000 ICI-treated patients (including 387 mUC patients) and identified clonal TMB as the strongest predictor of response overall, followed by total TMB, they found that clonal TMB and the APOBEC signature were among the most important features associated with response in a multivariable model predicting ICI response in bladder cancer.
Somatic alterations
Mutations and copy number alterations (CNAs) affecting specific genes have been linked to ICI outcomes.In their meta-analysis, Litchfieldet al.[95]identifiedTRAF2loss as a predictor of ICI response andCCND1amplification as a marker of ICI resistance both across cancer types and in UC specifically.Separately,biomarker analyses from clinical trials have found that somatic mutations in DDR or cycle regulator genes are associated with both TMB and response[105,106].
TRAF2 is a tumor necrosis factor (TNF) receptor-associated protein which inhibits TNF-mediated apoptosis[115].Interestingly,TRAF2loss had been independently identified in a CRISPR/Cas9 screen for genomic alterations sensitizing tumors to T cell killing[116].In the Litchfieldet al.[95]meta-analysis, loss of the 9q34 cytoband, and specifically theTRAF2locus within it, was the most significant CNA associated with ICI response (lost in 44% of respondersvs.30% of non-responders,P= 6.9 × 10-5).
CCND1encodes Cyclin D1, a cell cycle regulator that has also been shown to negatively regulate PD-L1 protein levels[117].In the Litchfield meta-analysis, UC was the cancer type with the mostCCND1amplification.Using various UC and pan-cancer clinical cohorts, they showed that higherCCND1expression levels were associated with lack of ICI response, andCCND1amplification was associated with worse OS among ICI-treated patients but was not associated with OS among non-ICI treated patients,suggesting a predictive relationship.However, they were unable to test for a formal interaction due to differences in the ICI and non-ICI treated cohorts[95].
Mutations in DDR pathway genes were associated with improved outcomes in exploratory analyses of both the IMvigor210 and JAVELIN Bladder 100 trials.However, in the IMvigor210 analysis, Mariathasanet al.[105]found that DDR mutations were not predictive of outcomes independent of TMB.This may also be the case in the JAVELIN study, but the biomarker analyses are currently only available in abstract form[86].
Gene expression
In contrast to somatic alterations in DNA, which are essentially fixed for each cell, gene expression is dynamic and reflects ongoing cellular processes at sampling.Gene expression profiling might therefore capture the state of the tumor and its microenvironment in a way that DNA sequencing does not.Several studies have used samples from mUC clinical trials to identify gene expression signatures correlated with response or resistance to ICI therapy[81,86,105,118].
Two broad categories of gene expression signatures have been linked to ICI response in prospective mUC cohorts: a group of genes reflecting cytotoxic T cell activity and a group of genes reflecting immunosuppressive stromal signaling.The former has been associated with ICI response, while the latter has been associated with ICI resistance.These signatures remain exploratory pending validation in additional prospective cohorts.
Gene expression analyses of data from TCGA, the CheckMate-275 trial of nivolumab, and the IMvigor210 trial of atezolizumab converged on the observation that certain types of stromal cell signaling contribute to an “immune-excluded” phenotype, wherein CD8 T cells are separated from tumor cells by dense connective tissue[105,119].Notably, TGFβ signaling can promote the differentiation of mesenchymal precursors into fibroblasts[120-122], and in IMvigor210 both a TGFβ ligand (TGFB1) and receptor (TGFBR2) were associated with non-response and reduced OS[105].Hypothesizing that TGFβ signaling in fibroblasts might be a biomarker for ICI response, Mariathasanet al.[105]showed that a pan-fibroblast TGFβ response signature (FTBRS) was associated with response in immune-excluded tumors.They further showed in murine tumor models that anti-TGFβ antibodies enhanced the efficacy of anti-PD-L1 therapy.A higher F-TBRS signature was also associated with worse OS for patients treated with atezolizumab rather than platinumchemotherapy in the IMvigor130 trial[110].
A variety of inflammatory gene signatures reflecting CD8 T cell activity and/or interferon-gamma signaling have been associated with ICI response in mUC[81,86,105,118].Some recurrent genes in these signatures include:CCL5,CD27,CD8A,CXCL9,CXCL10,CXCR6,GZMA,GZMB,IDO1,IFNG,LAG3,PRF1,STAT1, a n dTBX21.A 25-gene interferon-gamma signature was associated with response in the CheckMate-275 trial of nivolumab[81].An 8-gene subset of that signature focused on CD8 T effector activity was positively associated with response in IMvigor210[105].CXCL9andCXCL10expression were individually associated with ICI response in both IMvigor210 and CheckMate-275[81,105].Notably,CXCL9expression was one of the strongest predictors of ICI response in the Litchfieldet al.[95]meta-analysis of ICI biomarkers across tumor types.
Molecular subtypes
The TCGA molecular classification has been the most used schema in ICI clinical trials.The classification scheme, comprising 4 subtypes, was developed in 2014 using gene expression data from 131 muscle-invasive bladder tumors[65].It was updated in 2017 using an expanded cohort of 412 muscle-invasive specimens, with a fifth “neuronal” subtype added[36], though the analyses in mUC ICI trials have focused on the initial 2014 classification or have omitted the neuronal subtype.
Associations between molecular subtypes and ICI responses have been inconsistent across ICI trials and agents.Data from the IMvigor210 trial of atezolizumab found the highest response rates among the luminal II subtype[79,80,105], whereas the CheckMate-275 trial of nivolumab found the highest response among the basal I subtype[81].The JAVELIN Bladder 100 trial of maintenance avelumab found no association between subtypes and OS[123].
It may be that the molecular factors associated with ICI response do not segregate neatly into existing molecular classifications.In the analysis of IMvigor210, Mariathasanet al.[105]found that combining a separate molecular classification scheme (the Lund scheme) with the TCGA scheme could improve the separation of responders and non-responder.In this analysis, they found that about half of tumors classified as luminal II in the TCGA scheme could also be classified as genomically unstable (GU) according to the Lund scheme.Tumors that were both GU and luminal II had high TMB and low expression of the F-TBRS score associated with fibroblast TGFβ activity.On the other hand, tumors that were luminal II but not GU had low TMB, a high F-TBRS score, and had a significantly lower overall response rate[105].
Tumor microenvironment factors
Immune checkpoint inhibitors are not directly cytotoxic but rather alter immune cell signaling to promote immune-mediated destruction of tumor cells.Given this indirect mode of action, the factors that influence ICI outcomes extend beyond the tumor cells and include interactions between tumor, immune, and stromal cells within the tumor microenvironment (TME)[124,125].Thus far, we have discussed examples of both tumor-intrinsic (mutation burden, somatic alterations) and tumor-extrinsic (immune cell infiltration,fibroblast activity) candidate ICI biomarkers.Additional tumor-extrinsic ICI biomarkers may emerge as our understanding of the TME matures.
Early efforts to examine the TME in relation to ICI response used immunohistochemistry to correlate the spatial arrangement of tumor-infiltrating lymphocytes with outcomes[126].This gave rise to the paradigm of immune inflamed, immune cold, and immune excluded phenotypes, with immune inflamed tumors generally having better responses[105,126].Deconvolution algorithms have also been used to infer the immune composition of the TME from bulk tumor RNA[127,128]; these inferred cell populations can then be correlated with ICI outcomes[129].More recently, tools such as single-cell RNA-sequencing (scRNA-seq) and multiplex immunohistochemistry, are enabling higher-resolution analysis of the TME and its relationship to ICIresponse across cancer types[130,131].In contrast to bulk gene expression profiles, scRNA-seq enables the direct quantification and characterization of distinct cellular subpopulations.In urothelial carcinoma, this has been used to elucidate the heterogeneity of tumor, immune, and stromal cells[132-134].
Fibroblasts are of particular interest in mUC given the identification of a fibroblast-associated TGFβ signature as a predictor of atezolizumab response in the IMvigor210 analysis[105].Note that this analysis did not use scRNA-seq - the TGFβ signature was developedin vitroby exposing fibroblasts to TGFβ ligands and measuring changes in gene expression.The signature was then applied to bulk RNA-seq data as a proxy for TGFβ activity in fibroblasts.A separate single-cell analysis of 8 bladder cancer samples by Chenet al.[133]identified a subset of fibroblasts that were a major source of pro-proliferation growth factors and CXCL12 -a chemokine associated with the accumulation of immunosuppressive tumor-associated macrophages[133,135].However, TGFβ signaling in these fibroblasts was not reported.
Single-cell profiling may reveal novel determinants of ICI response which can be translated into biomarkers.For example, in a single-cell analysis of 7 bladder tumors, Ohet al.[132]identified cytotoxic CD4+T cell populations which had not been previously recognized in bladder cancer.A gene expression signature derived from these novel CD4 T cells was applied to bulk RNA sequencing data from the IMvigor210 study and was predictive of atezolizumab response among patients with an inflamed tumor microenvironment.
In another example combining bulk and single-cell analyses to dissect the UC TME, bulk gene expression data from two clinical trials were first used to identify two signatures associated with ICI response and resistance[136].The authors sought to identify the cell types responsible for each signature using single-cell data.Unexpectedly, they found that many cell types - and myeloid cells in particular - expressed a mix of genes from both signatures rather than being restricted to one signature.This led them to define a score based on the ratio of the two signatures within myeloid cells (Msc21R).Intriguingly, this single-cell expression-based score was associated with ICI outcomes when measured in the pre-treatment peripheral blood monocytes of 10 mUC patients.
While scRNA-seq remains limited to research settings for now, single-cell insights from the TME are likely to influence biomarker development moving forward.
TARGETED THERAPIES BIOMARKERS
Molecularly targeted therapies have also evolved over recent years, expanding the landscape of cancer treatment options available to patients.The target is often a mutated or overexpressed molecule with a critical role in tumor progression while being less important for normal cell function, creating a therapeutic window that can be exploited.In UC, current FDA-approved targeted therapies are the FGFR inhibitor erdafitinib, the Nectin-4 ADC EV, and the Trop-2 ADC SG.
Erdafitinib, a pan-FGFR-kinase inhibitor, was granted accelerated approval in 2019 for patients with locally advanced or metastatic UC with progression after platinum-based chemotherapy and known susceptibleFGFR2/3alterations.The specific alterations includeFGFR3mutations (R248C, S249C, G370C, Y373C) orFGFR2/3gene fusions (FGFR2-BICC1, FGFR2-CASP7, FGFR3-TACC3, FGFR3-BAIAP2L1).Accelerated approval was based on a phase II trial demonstrating overall response rates of 30%-40% in this biomarkerdriven population[7,137].The confirmatory phase III THOR trial (NCT03390504) is currently underway,directly comparing erdafitinib to pembrolizumab or single-agent chemotherapy in previously treated mUC.
EV is an ADC consisting of a monoclonal antibody specific for Nectin-4 conjugated to monomethyl auristatin E (MMAE), a microtubule-disrupting agent[138,139].Accelerated approval was granted for locally advanced or metastatic UC after prior platinum-based chemotherapy and ICI as a result of phase II EV-201 trial.In the confirmatory phase III EV-301 trial, EV conferred a significant survival benefit over standard chemotherapy in the post-chemo/post-ICI setting, leading to regular FDA approval[4].Notably, EV has shown benefit and is approved for treatment without regard to Nectin-4 levels.
The third targeted therapy approved in mUC is SG, a monoclonal antibody specific for Trop-2 conjugated with SN-38, the active metabolite of irinotecan[140].Accelerated approval for SG was recently granted in April 2021 after the phase II TROPHY-U-01 trial demonstrated a 27% ORR and 10.9 months median OS in the post-chemo/post-ICI setting[5].Similar to EV, SG has been tested and approved without regard to the levels of its target, Trop-2.
Several other targets are under investigation in UC.Below we describe approved and prospective targeted therapies in more detail, with an emphasis on biomarkers that may guide treatment options.
FGFR
The FGFR proteins comprise a family of four transmembrane receptor tyrosine kinases, FGFR 1-4, with roles in cellular proliferation, differentiation, apoptosis, migration, and angiogenesis[141,142].Upon binding to their ligand, these receptors undergo dimerization and autophosphorylation resulting in subsequent activation of the RAS-MAPK and PI3K signaling pathways.Dysregulation of theFGFRpathway has been linked to oncogenesis in multiple cancer types, including UC[143].FGFR3alterations are observed in approximately 81% of non-invasive UC and up to 54% of invasive UC[144], and are more commonly seen in the luminal-papillary molecular subtype that comprises 35% of MIBC[36].The most commonFGFR3mutation is S249C (61% ofFGFR3-mutated UC), followed by Y375 (19%), R248C (8%), and G370C (6%), all conferring downstream signaling activation through ligand-independent receptor dimerization or constitutive receptor activity[145].Amplification of the FGFR genes occurs in UC with a prevalence of approximately 7% forFGFR1and 2% forFGFR3[146].Finally,FGFRfusion pairings also lead to signaling activation, most notable betweenFGFR3and fusion partnerTACC3in approximately 2.6% of UC[65,147].
As described above, erdafitinib has been approved for use in previously treated mUC with susceptibleFGF2/3alterations.It remains to be seen whether erdafitinib may benefit patients in combination with other therapies or in earlier lines of therapy.The phase Ib/II NORSE trial (NCT03473743) may answer some of these questions, evaluating erdafitinib in combination with cetrelimab (anti-PD-1) and platinum chemotherapy inFGFR-altered patients with or without prior treatment and in treatment-naïve cisplatinineligible patients[148].
Multiple additional FGFR inhibitors have also been evaluated in UC.Here we highlight notable results and differences between them.Infigratinib (BGJ398), an FGFR1-3 inhibitor, was tested in patients with the sameFGFRalterations as erdafitinib, as well as Y375C, K652M/T, K652E/Q[149], and is currently being studied in phase III PROOF302 trial (NCT04197986) as adjuvant therapy for cisplatin-ineligible bladder cancer or upper tract UC.
Vofatamab (B-701), a monoclonal antibody that inhibits FGFR3, is being tested inFGFR3-altered mUC in the post-chemotherapy setting in the phase Ib/II FIERCE-21 trial[150,151].Of note, the phase Ib/II FIERCE-22 trial found promising response rates for vofatamab in combination with pembrolizumab in bothFGFR3-mutant (ORR 43%) andFGFR3-wild-type (ORR 33%) patients[152].Vofatamab is known to inhibit wild-typeFGFR3, which may play a part in reversing the ICI-insensitivity attributed to FGFR signaling[152].
Derazantinib (ARQ087) is an FGFR2 selective inhibitor in the phase Ib/II FIDES-02 trial as monotherapy or in combination with atezolizumab for cisplatin-ineligible advanced UC withFGFR1-3mutations and fusions.The inhibitor also has known immunomodulatory activity on macrophages via CSF1R[153,154].
Additional FGFR inhibitors include Debio 1347, an FGFR1-3 inhibitor being tested only forFGFR1-3fusions[155], TAS-120, an FGFR1-4 inhibitor under evaluation for FGFR alterations that include amplifications[156], pemigatinib, an FGFR1-2 inhibitor being studied for NMIBC with recurrent low or intermediate risk disease, and, dovitinib, an FGFR1-3 inhibitor which demonstrated limited efficacy[157].
FGFR1-3mRNA overexpression has also been tested as a biomarker, with equivocal results.Among patients with increasedFGFR1/3mRNA levels, rogaratinib demonstrated similar outcomes as chemotherapy in the post-platinum setting in phase II/III FORT-1 trial.However, the response rate was highest among those withFGFR3somatic alterations, suggesting that mRNA overexpression may be a less selective biomarker than bona fide somatic alterations[158].Conversely, the phase Ib/II FORT-2 trial reported favorable responses for rogaratinib in combination with atezolizumab amongFGFR1-3mRNA overexpressing mUC patients regardless ofFGFRDNA alterations or PD-L1 level, though the sample size was limited in this early-phase trial[159].The utility ofFGFRmRNA overexpression remains unclear, especially in the setting of FGFR inhibitor monotherapy.
Finally, ctDNA may serve as both a prognostic and treatment response biomarker for FGFR inhibitors.In the BISCAY trial, which tested durvalumab in combination with targeted therapies in biomarker-selected populations,FGFRalterations identified in tissue samples could be accurately detected in ctDNA.Higher baseline ctDNA levels correlated with worse OS.Furthermore,FGFRalterations in ctDNA declined with response to therapy and increased with clinical progression[91].
It is also important to pinpoint markers of treatment resistance, either at baseline or acquired after therapy,that may aid in clinical decision making.There are multiple gatekeeper mutations located in the FGFR ATP-binding pocket known to confer resistance to FGFR inhibitors[160].In particular, infigratinib has a 5-10-fold decreased efficacy with the K650E gatekeeper mutation[161].Additionally, there are multiple secondaryFGFR2mutations are identified as sources of resistance, such as V564F after infigratinib treatment[162].While these tumor cells appear to maintain dependence on the FGFR signaling pathway,activation of other pathways such as PI3K-AKT and RAS-MAPK also serve as mechanisms of resistance[160,163,164].Lysosome-mediated TKI sequestration, activating gene fusions, and epithelialmesenchymal transition are further mechanisms that limit FGFR inhibitor efficacy[160,165].These markers may serve to predict non-responders to FGFR inhibition, identify specific inhibitors that may be more efficacious for an individual, or guide discontinuation of FGFR-targeted therapy.
Nectin-4
Enfortumab vedotin is an ADC targeting Nectin-4, a transmembrane protein involved in cellular adhesion and upregulated in UC with limited expression in benign tissue[166].Despite the targeted nature of EV,Nectin-4 is not currently utilized as a biomarker in patient selection.The EV-101 phase I trial initially required Nectin-4 expression for inclusion, but the protocol was later amended to remove that requirement because nearly all screened tumors expressed Nectin-4 at high levels[167].Subsequently, the pivotal phase III EV-301 trial demonstrated an OS benefit for EV in a biomarker-unselected population[4].
Nevertheless, emerging treatment options in the same clinical setting (e.g., sacituzumab govitecan) provide an impetus to identify EV biomarkers for treatment selection and prioritization.In a recent abstract,NECTIN4gene expression was found to vary by molecular subtype and was enriched in luminal-type UC.In both luminal and basal bladder cancer cell lines, overexpressing and knocking outNECTIN4conferred sensitivity and resistance, respectively, to EVin vitro[168].Nectin-4 protein levels vary by disease stage (87%NMIBCvs.58% MIBC) and by morphology (e.g., 28% in micropapillary tumors, 63% in plasmacytoid)[169].It remains to be seen whether Nectin-4 expression at the RNA or protein level is associated with clinical outcomes or may interact with molecular subtype or variant histology.
Ongoing trials with EV include the phase Ib/II EV-103/KEYNOTE-869 trial (NCT03288545) evaluating the ADC in combination with pembrolizumab and/or chemotherapy for MIBC, locally-advanced, and metastatic UC[170], with preliminary analysis showing promising results in combination with pembrolizumab as first-line therapy for cisplatin-ineligible patients[171].EV-302 (NCT04223856) is evaluating EV with pembrolizumab compared to standard of care chemotherapy for untreated locally advanced or metastatic UC, NCT04878029 is looking at EV with cabozantinib for advanced UC, and NCT04724018 is testing EV with SG post-chemo and ICI, all of which will not be biomarker-driven.
Trop-2
Trop-2 is a transmembrane glycoprotein involved in calcium signaling and cellular proliferation, with reported overexpression in up to 83% of UC, and, importantly, limited expression in normal tissues[172,173].High Trop-2 expression has also been associated with more advanced disease and recurrence[174].Similar to EV, SG was studied and approved in a biomarker-independent manner.
As a biomarker, Trop-2 overexpression in other malignancies has been paradoxically linked to growth promotion after oxaliplatin[140], and is associated with cisplatin resistance[175]and response to AKT inhibitors[176].In breast cancer, Trop-2 expression is differentially expressed among hormone receptorpositive and triple-negative disease[177], and biomarker analysis of a phase III trial demonstrated that while SG benefitted patients regardless of Trop-2 expression, the benefit was greatest among the high Trop-2 expression subgroup[178].As more options become available in the post-chemo/post-ICI setting, biomarker selection will become increasingly important, and more intricate analysis of mRNA expression and molecular subtypes may also prove meaningful for Trop-2.
Ongoing SG trials include the phase III TROPiCS-04 trial evaluating SG for advanced UC progressing after platinum-based chemotherapy and ICI[179], and the phase Ib/II umbrella trial with SG in combination with atezolizumab for cisplatin-ineligible MIBC and platinum-refractory advanced UC[180].
ErbB (Her2, EGFR)
Human epidermal growth factor receptor 2 (HER2/ErbB2) is a cancer biomarker with approved targeted therapies for positive breast and gastroesophageal cancer patients.The protein is one of four in a family of receptors with tyrosine kinase activity involved in cellular proliferation and differentiation[181], with overexpression prevalence in UC cohorts as high as 76%[182,183].
Multiple Her-2 inhibitors have been tested in UC, with limited success.Trastuzumab initially showed promising results for advanced chemo-naive UC in combination with platinum-based chemotherapy[184],but a subsequent trial showed no added benefit[185].Lapatinib, a Her-2 and EGFR pathway inhibitor, also failed to improve outcomes in a phase III trial for HER1/2-positive mUC after first-line chemotherapy[186].
Conversely, afatinib, a Her-2/4 and EGFR inhibitor, has demonstrated biomarker-driven potential for platinum-refractory mUC.PFS at three months was 83% in patients withHER2,EGFR,ERBB3, orERBB4alterations, compared to 0% of patients without these alterations[187].The follow-up phase II LUX-Bladder 1 trial assessing afatinib forERBB1-3altered mUC in the post-platinum setting is ongoing[188].Additionally,the basket trials My Pathway (NCT02091141)[189]and NCI-MATCH (NCT02465060) are currently evaluating trastuzumab and pertuzumab in bladder cancer withHER2overexpression or amplification.
Multiple ADCs targeting Her-2 are also being tested in UC.Disitamab vedotin (RC48-ADC), a Her-2 antibody conjugated with MMAE, demonstrated promising responses for advanced UC after at least 1 prior line of therapy[190].Trastuzumab deruxtecan (DS-8201), an ADC pairing trastuzumab with a topoisomerase I inhibitor, is in an ongoing phase Ib trial (NCT03523572) in combination with nivolumab for Her-2 positive mUC after progression on platinum-based chemotherapy, and the basket trial NCT02675829 is testing adotrastuzumab emtansine (TDM-1) in aHER2-amplified bladder cancer cohort[191].
Further trial data may elucidate the optimal utilization of Her-2 as a biomarker for either Her-2 inhibitors or ADCs.Gene expression, gene amplification, and protein level by IHC, FISH, or serum levels may all play a role in treatment and patient selection.Molecular subtypes may also be useful in guiding EGFR-based therapy, as basal-like tumors demonstrated increased EGFR pathway activity and sensitivity to EGFR-based treatment in pre-clinical models[69].
PI3K/AKT/mTOR, MAPK pathway
The PI3K/AKT/mTOR pathway serves to promote cellular growth, proliferation, and angiogenesis, and regulates apoptosis and autophagy.PI3K is a lipid kinase activated via phosphorylation by RTKs and GPCRs upon ligand binding.The protein then phosphorylates and converts PIP2 into PIP3, a process inhibited via dephosphorylation by the tumor suppressor PTEN.PIP3 subsequently recruits the kinase AKT, which functions to inhibit apoptosis through Bcl-2 and activates mTOR through suppression of TSC1 and TSC2[192].Genomic alterations throughout this pathway result in increased downstream activity and are common in many malignancies.Aberrations in the pathway are seen in up to 72% of UC[193], commonly throughPIK3CAmutations (21%-25% of MIBC)[65,194,195], loss ofPTENexpression (39%-94%)[195-199], loss of heterozygosity ofTSC1(40%-50%) orTSC2(15%)[195,200], or due to aberrations in the upstream FGFR or ERBB pathways as outlined above.
Everolimus, an mTOR inhibitor, has been tested in the post-chemotherapy setting for UC[201].Notably, one patient with an exceptional response was found to have a loss-of-function mutation inTSC1, generating significant interest in mTOR pathway mutations as biomarkers for everolimus response.When everolimus was tested in combination with the TKI pazopanib,TSC1,TSC2, ormTORmutations were identified in the only complete responder (CR), and two of the three partial responders[202], and subsequent analysis of the CR revealed two distinct activating mTOR mutations, both validatedin vitroand conferring sensitivity to mTOR inhibition[203].However, follow-up studies have failed to yield clinically relevant efficacy for mTOR inhibition in UC treatment, including additional inhibitors such as sapanisertib, which showed minimal response forTSC1/2-mutated mUC patients[204].
The PI3K inhibitor, buparlisib (BKM-120), was also evaluated in the post-platinum setting, and, similarly,analysis of multiple responders revealedTSC1orPIK3CAmutations.However, an expansion cohort for patients with PI3K pathway alterations was terminated after no patients achieved disease control[205].Copanlisib and GSK2636771 are additional PI3K inhibitors currently under investigation in the basket NCIMATCH trial (NCT02465060) for patients withPI3KCA-activating mutations or loss/mutations ofPTEN[206].
AKT inhibitors (MK-2206, AZ7328) have also been tested in pre-clinical studies, with sensitivity dependent onPIK3CAmutations and resistance seen withPTEN,TSC1, andRASalterations[207,208].Further, dual PI3K and mTOR inhibitors, such as dactolisib (BEZ235) and omipalisib (GSK2126458), have also undergone evaluation for UC[209,210].
PARP/DNA repair
PARP genes can detect DNA damage and subsequently recruit downstream proteins involved in DNA repair.The PARP inhibitors olaparib, rucaparib, niraparib, and talazoparib have gained FDA approvals,primarily as monotherapy for ovarian, breast, prostate, and pancreatic cancer with germline or somaticBRCA1/2mutations, owing to the synergistic synthetic lethality in tumors with HRR deficits[211].BRCA1/2mutations occur in approximately 1.5% and 1.4%, respectively, of UC[49].
Rucaparib was tested in UC in the phase II ATLAS trial, with no observed relationship between HRR deficiency and efficacy; only 4 of the 97 patients hadBRCA1/2deficiencies[212].Ongoing phase II trials are evaluating olaparib in mUC with at least one somaticDDRgene alteration (NCT03448718, NCT03375307).Olaparib did not appear to confer additional benefit when combined with durvalumab in the phase Ib BISCAY study (NCT02546661), even among those with HRR mutations[91].The combination is also being evaluated for platinum-ineligible mUC in phase II BAYOU trial (NCT03459846).
It remains to be seen whether PARP inhibition in HRR-deficient patients will prove efficacious for UC.However, as outlined above, various DNA repair genes have potential as predictive biomarkers for chemotherapy response, and chemotherapy itself is also involved in DNA repair dysregulation, raising the possibility of synergistic efficacy with PARP inhibition.
Additional TKIs/VEGF inhibitors
Multiple non-specific TKIs have been evaluated for UC.Sorafenib, a multi-targeted TKI acting on the VEGF, RAF/MEK/ERK, and PDGF pathways, was tested in the neoadjuvant setting in combination with cisplatin/gemcitabine.Responders demonstrated higher rates of mutations in DNA-repair genes, RAS-RAF pathway genes, chromatin-remodeling genes, and HER-family genes (of note only HER-family genes were statistically significant given the study’s limited power, 36.4%vs.0%)[24].The TKI is also under evaluation for post-platinum mUC.
Cabozantinib, which has immunomodulatory properties involving Tregs and myeloid-derived suppressor cells, has also been tested in platinum-refractory mUC.Biomarker analyses have suggested that cabozantinib may counteract an immunosuppressive TME, providing the rationale for its combination with ICIs[213].A combinatorial phase I trial testing cabozantinib with nivolumab and/or ipilimumab has shown promising results, with baseline circulating tumor cell subsets (defined by EpCAM, MET, and CXCR4 expression) associated with median OS[214].Ongoing trials such as ARCADIA (NCT03824691) and COSMIC-021 (NCT03170960) are testing cabozantinib-ICI combinations with plans for extensive correlative studies to support biomarker development, including immunophenotyping, gene expression profiling, and somatic mutation testing in both tumor and plasma[215,216].
Other recently reported trials of targeted therapies with the potential for biomarker analysis include the RANGE study of ramucirumab with docetaxel (NCT02426125), and the multi-cohort study of sitravatinib in mUC (NCT03606174).The RANGE study found that ramucirumab (a monoclonal antibody targetingVEGFR-2) with docetaxel improved PFS but not OS among previously treated mUC patients[217].However,high PD-L1 expression was associated with improved PFS, and further biomarker analyses may uncover patient subsets who derive more benefit.Sitravatinib, another multi-targeted TKI with immunomodulatory activity, is being tested in combination with nivolumab, and biomarker analyses are planned[218].
Multi-targeted TKIs are an intriguing future prospect for mUC management.Many have immunomodulatory properties, which make them promising candidates for ICI combinations.However,their pleiotropic effects through multiple pathways may mask the patient subsets who derive the most benefit, making biomarker analyses even more essential.
CONCLUSION
There are now three distinct therapeutic classes for the treatment of metastatic urothelial cancer:chemotherapy, immunotherapy, and targeted therapies [Figure 3].This presents medical oncologists with the challenge of finding the optimal sequence and combination of these agents to maximize outcomes for their patients.The precision medicine paradigm aims to divide urothelial cancer into increasingly specific subtypes, each with its own optimal treatment plan.However, realizing this vision requires both a variety of effective treatment options and reliable biomarkers to distinguish the relevant disease states.As it stands,biomarker development has lagged in therapeutic development, with only PD-L1 expression andFGFRalterations playing a routine role in treatment selection.
Nevertheless, there is reason to be hopeful that greater resolution of biomarker-defined subgroups will be achieved.Research into the basic mechanisms underlying urothelial cancer and its response to therapy has already yielded various candidate biomarkers, as detailed in this review.More candidate biomarkers will be identified as researchers use increasingly precise and comprehensive molecular assays to interrogate tumor biology, such as single-cell RNA sequencing and untargeted proteomics.Clinical molecular assays are also becoming more advanced.Sequencing panels are including more genes and may ultimately include the whole exome[219].Commercial transcriptomic tests are being introduced into the clinic[220].Notably,circulating tumor DNA is routinely sequenced in other cancers and has shown promise as a biomarker forboth treatment response and prognosis in mUC.These practical advances, as much as basic and translational developments, are essential to realizing precision medicine in the clinic.
However, there are also reasons to be circumspect.Tumor biology is complex, and there may be a limit to the resolution that can be achieved by biomarkers based on a single marker or on a single type of biological data.However, more complex biomarkers may be challenging to deploy clinically.Certain investigative biomarkers utilize novel methods (e.g., single-cell sequencing) that may not be accessible to clinicians,though it is sometimes possible to derive related biomarkers using clinically available methods.Finally,biomarker discovery and validation require large amounts of curated data, which in turn requires significant coordination and foresight to accrue.Several databases and consortia have attempted to meet this challenge,but in general, these public efforts have not reached the necessary scale.More work and resources are needed to accelerate biomarker discovery, development, and translation.
In conclusion, the accelerating progress in therapies for metastatic urothelial cancer necessitates greater investment in biomarker discovery and validation to enable optimal treatment for bladder cancer patients.
DECLARATIONS
Acknowledgement
The authors gratefully acknowledge Jill Gregory for her medical illustrations.
Authors’ contributions
Drafting of the manuscript: Jun T, Anker J Review, editing, and supervision: Galsky MD
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
Tomi Jun is employed by Sema4.Matthew Galsky has received research funding from Bristol Myers Squibb,Novartis, Dendreon, Astra Zeneca, Merck, Genentech; has consultant or advisory board roles for Bristol Myers Squibb, Merck, Genentech, AstraZeneca, Pfizer, EMD Serono, Seattle Genetics, Janssen, Numab,Dragonfly, GlaxoSmithKline, Basilea, UroGen, and Rappta Therapeutics.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2022.
 Journal of Cancer Metastasis and Treatment2022年1期
Journal of Cancer Metastasis and Treatment2022年1期
- Journal of Cancer Metastasis and Treatment的其它文章
- New developments in management of metastatic thyroid cancer
- AUTHOR INSTRUCTIONS
- The role of radiotherapy in metastatic bladder cancer
- Serum squamous cell carcinoma antigen is a predictive factor of outcomes in patients with locally advanced unresectable esophageal squamous cell carcinoma treated by definitive chemoradiotherapy
- The role of MT1-MMP in the progression and metastasis of osteosarcoma