
基于比较转录组学的多花黄精黄酮类化合物合成基因表达分析
2022-04-29 09:57叶碧欢朱杰丽石从广陈友吾胡传久宋其岩李海波
食品与生物技术学报 2022年4期
叶碧欢, 杨 阳, 朱杰丽, 石从广,陈友吾, 胡传久, 宋其岩, 李海波*
(1. 浙江省林业科学研究院,浙江 杭州 310023;2. 浙江农林大学 林业与生物技术学院,浙江 杭州 311300)
黄精属 Polygonatum Mill. 隶属百合科(Liliaceae),为多年生草本植物,在世界范围内广泛分布。 该属植物在我国有31 种,除南方热带以外,南北方各有分布[1]。 在黄精属植物的化学成分和药理活性研究方面,多集中于甾体皂苷和多糖二种主要活性成分[2-3],对黄酮类化合物的报道较少且不深入,大多为总黄酮提取工艺、含量测定以及生物活性评价[4-6]。 通过比较《中国药典》记载的3 种黄精新鲜块茎的总黄酮质量分数显示, 黄精P. sibiricum Red.、滇黄精P. kingianum Coll. et Hemsl.和多花黄精P. cyrtonema Hua. 的总黄酮质量分数均比较低,分别为0.018~0.035、0.015~0.030、0.004~0.034 mg/g,且在不同黄精属种间以及不同产地间存在很大差异[7]。
黄酮类化合物是一类广泛分布于植物体内的次生代谢物,目前已从黄精属8 种植物中共分离出6 种(高异黄酮类、异黄酮类、黄酮类、查耳酮类、二氢黄酮类、紫檀烷类)不同结构亚型的化合物54 个[8],其中高异黄酮类(Homoisoflavones)最多,是该属植物的特征性成分,在自然界颇为少见,有降血糖、抗炎、抗氧化、抗肿瘤、抗衰老等药理活性,具有重要的研究价值[9-10]。因此,研究黄精属植物中黄酮类,特别是高异黄酮类化合物的生物合成途径与调控机制, 可为黄酮类化合物的合成生物学研究奠定基础,也为通过外源诱导手段促进黄酮类物质的大量积累,以及今后黄酮类药物和保健品的产业化研发开辟新的来源途径。
近年来,转录组测序技术已广泛用于药用植物新基因的发现、分子标记挖掘、代谢途径的确定等研究,有助于次生代谢相关基因的挖掘与功能研究[11-16]。迄今,黄精属植物的转录组学研究多在黄精和滇黄精2 个物种上开展,且以多糖和甾体皂苷为主[17-21],在多花黄精物种上仅有多糖合成相关基因的报道[20],有关黄精属植物黄酮类化合物生物合成途径的分析与相关基因鉴定尚属空白。
作者将对多花黄精组培苗不同发育期的根茎进行比较转录组学研究,拟基于转录组数据的组装与注释,鉴定与黄酮类化合物生物合成相关的关键基因,并对在多花黄精幼苗期根茎中表达的转录因子(Transcription factor,TF)进行鉴定与分类。基于转录组FPKM 数据筛选块茎不同发育期的差异表达基因, 分析差异表达基因在次生代谢通路的富集,比较黄酮类化合物生物合成关键基因在不同发育期块茎中的差异表达, 并进行实时荧光定量PCR(RT-qPCR)验证。 本研究可弥补黄精属植物黄酮类化合物生物合成途径基因资源的空白,为后续研究黄酮类化合物合成与积累的分子调控机制奠定基础。
1 材料与方法
1.1 材料来源
以多花黄精种子作为外植体,经消毒处理后无菌诱导其植株再生,建立组培快繁体系。 组培培养基为1/2 MS(Murashige & Skoog)。 培养条件为温度25 ℃,湿度50%,光照2500200 lx。 以培养于浙江省林业科学研究院林木育种组培实验室的多花黄精Polygonatum cyrtonema 组培苗为试材。 从组培瓶中取出分别培养了3 个月和9 个月的多花黄精幼苗,切去茎叶,取带芽根茎立即用液氮速冻,存储于-80 ℃作为转录组学研究用材料。 两种材料均取6 瓶, 其中3 瓶以去除须根后的块茎作为总黄酮质量分数分析的3 个生物学重复,另3 瓶作为转录组测序的3 个生物学重复。
1.2 总黄酮质量分数测定
多花黄精块茎总黄酮的测定参考文献[22]的方法,以芦丁作为对照品,利用紫外分光光度计测定各样品510 nm 处的吸光度(A510nm)。 以芦丁质量浓度为横坐标,吸光度为纵坐标,绘制出标准曲线,得到回归方程。 多花黄精块茎总黄酮的提取参考文献[23]的方法并略微修改,即称取完全干燥粉碎的块茎10 g,加入甲醇500 mL,于40 ℃下超声提取1 h,过滤,取滤液60 mL,加入100 mL 酸解液(75 mL 甲醇+25 mL 盐酸),水浴回流酸解100 min,加甲醇定容,过滤,取滤液检测A510nm。 根据芦丁标准曲线和回归方程,计算不同样品的总黄酮质量分数。

式中:I 为总黄酮质量分数,mg/g;C 为样品提取液的总黄酮质量浓度,mg/mL;N 稀释倍数;V 为定容体积,mL;m 为多花黄精块茎样品质量,g。
1.3 转录组测序与序列组装和功能注释
转录组测序与序列组装和功能注释的具体方法参考作者所在课题组前期研究报告[24],简述为:采用Plant Total RNA Miniprep Purification Kit(TR02,GeneMark,Taiwan,China) 提取多花黄精根茎的总RNA。 采用SureSelect Strand-Specific RNA Library Prep for Illumina Multiplexed Sequencing (Agilent Technologies,G9691)构建测序文库;使用Trinity 软件(v2012-10-15)对测序得到的高质量clean reads从头组装, 再利用CAP3 (r12/21/07) 软件拼接为unigene; 将unigene 序 列 比 对 到Nr、SwissProt、KEGG、COG/KOG 和Pfam 蛋白数据库以及拟南芥基因组数据库TAIR10(E-value≤1×10-5),得到与给定unigene 具有最高序列相似的蛋白质, 获得蛋白质功能注释信息。
1.4 转录因子鉴定
利 用Blast 程 序(https://www.ncbi.nlm.nih.gov/)将unigene 序列与Nr 蛋白数据库中的转录因子信息进行相似性比对(阈值:E-value≤1×10-5),得到与给定unigene 具有最高序列相似性的TF, 获得TF注释信息。
1.5 差异表达基因的筛选与功能富集分析
采 用 Cuffdiff (v 2.1.1) 软 件 计 算 FPKM(Fragments per kilobase of transcript per million fragments mapped)值[25]作为衡量转录本表达水平的指标,使用错误发现率FDR(False discovery rate)进行可信度检验。 差异表达基因的筛选标准为:倍数差异(Fold change,FC)≥2,FDR<0.01。 根据基因在不同样品中的表达量进行差异表达分析与功能富集分析, 差异表达基因的KEGG 富集分析采用KOBAS(v 2. 0)软件。 基于KEGG 注释的代谢途径统计与次生代谢相关的unigene 数量, 基于Nr 和TAIR10 注释信息统计与黄酮生物合成相关的unigene 数量。根据注释结果鉴定参与黄酮类化合物生物合成途径的unigene 及其编码的代谢酶。
1.6 RT-qPCR 分析基因表达
取上述1.3 中用于转录组测序的RNA 样品进行RT-qPCR 分析。 以RNA 为模板使用Prime ScriptTMRT reagent Kit with gDNA Eraser Kit(Perfect Real Time)逆转录合成cDNA 第一链,逆转录方法参照试剂盒说明。 用于RT-qPCR 分析的内参基因为UBQ-E2-10 和EF-1α2。 扩增目的基因和内参基因的引物序列见2.1 部分。 使用Oligo 7.57 软件设计特异性引物,引物合成由杭州有康生物科技有限公司完成。
RT-qPCR 分析参考作者所在课题组前期研究报告[26],具体为:反应在LineGene 9600 Plus 上进行。 反应体系为(10 μL):cDNA 模板1 μL,上、下游引 物 均 为0.2 μL,2×SYBR Green Mix (BioEasy Master Mix)5 μL,ddH2O 3.6 μL。 扩增程序为:恒温段95 ℃2 min;循环段95 ℃15 s,60 ℃20 s,40 个循环;溶解段95 ℃15 s,60 ℃1 min,95 ℃15 s。 每个实验设3 个技术重复。
采用2-△△Ct法[27]计算RT-qPCR 实验中的基因相对表达量。
2 结果与分析
2.1 多花黄精转录组测序结果
基于Illumina HiSeq 2500 测序平台获得多花黄精3 个月和9 个月幼苗期根茎的转录组数据。 经对原始数据进行统计, 去除raw data 中的接头序列及低质量reads,共获得约12 GB 的高质量clean data。通过对2 个时期转录本数据的合并组装,经拼接后共得到73218 条unigenes 序列(≥300 bp),其中长度在1 kb 以上的有17249 条。 所有unigenes 序列与各大数据库进行注释比对, 结果显示, 共有35 511 条unigenes 获得基因注释信息, 占全部unigenes 的48.5%, 其中长度大于1 kb 以上的有16640 条(见表1)。

表1 数据库注释的unigene 数量Table 1 Annotated unigene numbers in databases
2.2 黄酮类化合物生物合成相关酶基因鉴定
根据对2 个不同发育期根茎样品在各大数据库的整体注释,共鉴定出与黄酮类化合物生物合成相关的unigenes 83 条,编码18 个代谢酶(见表2)。目前,异黄酮类化合物(Isoflavones)的生物合成途径比较明确, 涉及的关键酶包括PAL、CHS、CHI 和异黄酮合成酶(IFS)[28-31]。 本研究在鉴定出的83 个酶基因中包括了PAL、CHS、CHI、F3H、FLS、DFR、F3′,5′H 等一批黄酮类化合物生物合成的关键性酶基因,但IFS 酶基因没有得到注释,这可能是由于在培养了3~9 个月的多花黄精幼苗期,其根茎中IFS 基因尚未充分表达,也可能与unigenes 的组装拼接质量有关。

表2 多花黄精转录组黄酮类化合物合成途径中的酶基因Table 2 Genes involved in the synthetic pathway of flavonoids in P. cyrtonema transcriptome
2.3 转录因子分析
转录因子是在转录水平上调控结构基因表达的关键因素之一,在调节植物生长发育、活性成分合成和环境胁迫中的基因表达发挥了主要作用。 许多转录因子家族由不同的基因家族组成, 包括bHLH、ERF、MYB-relaed、C2H2、NAC(NAM,ATAF1/2,CUC1/2)等。转录组数据显示,在多花黄精幼苗期的根茎中,共有1359 个编码转录因子的基因表达,可以分类为56 个TF 家族(见图1),以bHLH 类型最丰富(94 个,占比6.92%),其次是ERF(82 个,占比6.03%),MYB-relaed(79 个,占比5.81%)、C2H2(67个,占比4.93%)和NAC(58 个,占比4.27%)。MYBrelaed 和bHLH 转录因子单独或协作调节类黄酮次生代谢生物合成途径中结构基因的表达,从而控制植物体内类黄酮的生物合成[32]。 利用bHLH 转录因子调节药用植物活性物质生物合成的研究备受关注,其对黄酮类物质(花青素等)、生物碱(尼古丁等)、 萜类等活性成分生物合成的调控作用已被证实[33]。 本研究中鉴定的转录因子为今后研究多花黄精活性成分(多糖、皂苷和黄酮类化合物等)的积累与外源调控机制提供了丰富的基础数据。

图1 多花黄精转录组中转录因子的分布Fig. 1 Distribution of transcription factors in P.cyrtonema transcriptome
2.4 不同发育期差异表达基因的筛选与分析
筛选多花黄精2 个不同发育期根茎样品间表达水平显著差异的基因,形成差异基因数据库。 通过分析差异基因数据库共获得2602 个差异基因,占总被注释unigene 的7.33%,其中上调基因1866个,下调基因736 个(见图2)。这些差异表达基因为后续开展次生代谢通路分析提供了基础数据。
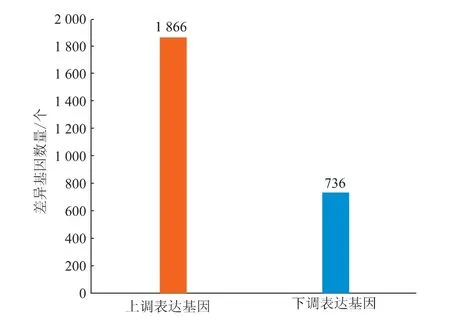
图2 多花黄精不同发育期块茎的差异表达基因Fig. 2 Differentially expressed genes in P. cyrtonema rhizome at different development stages
2.5 差异表达基因的KEGG 通路富集分析
为了进一步了解2 个不同发育期根茎样品间差异表达基因参与的代谢通路,对这些差异表达基因进一步利用KOBAS 进行KEGG 富集分析, 发现有497 条unigenes 注释到了109 个代谢通路中,且主要富集在代谢组(Metabolism)和遗传信息处理组(Genetic information processing)。 其中富集的前20条通路统计如图3 所示。
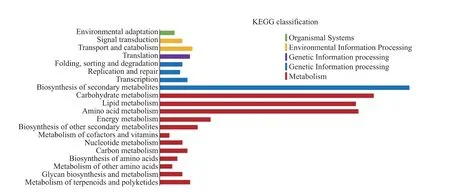
图3 差异表达基因的KEGG 注释分类Fig. 3 KEGG classification for differentially expressed genes in P. cyrtonema rhizome
在497 条unigenes 富 集 的109 个KEGG 代 谢通路中, 其中186 条定位到了19 个次生代谢物生物合成通路中,数量最多的是参与次生代谢物合成(Biosynthesis of secondary metabolites)通路,共118条,最少的是咖啡碱代谢(Caffeine metabolism)、吲哚类生物碱合成(Indole alkaloid biosynthesis)、黄酮和黄酮醇类合成(Flavone and flavonol biosynthesis)和硫代葡萄糖苷合成(Glucosinolate biosynthesis)通路,都仅只有1 条(见表3)。 此外,这些差异表达的酶基因也参与了像二苯乙烯类(Stilbenoids)、苯丙素(Phenylpropanoids)、萜 类(Terpenoids)、托 品 烷 类(Tropanes)、类胡萝卜素(Carotenoids)、油菜素甾醇(Brassinosteroids)、 黄 酮 类 (Flavonoids) 和 甾 体(Steroids)等次生代谢物的合成。
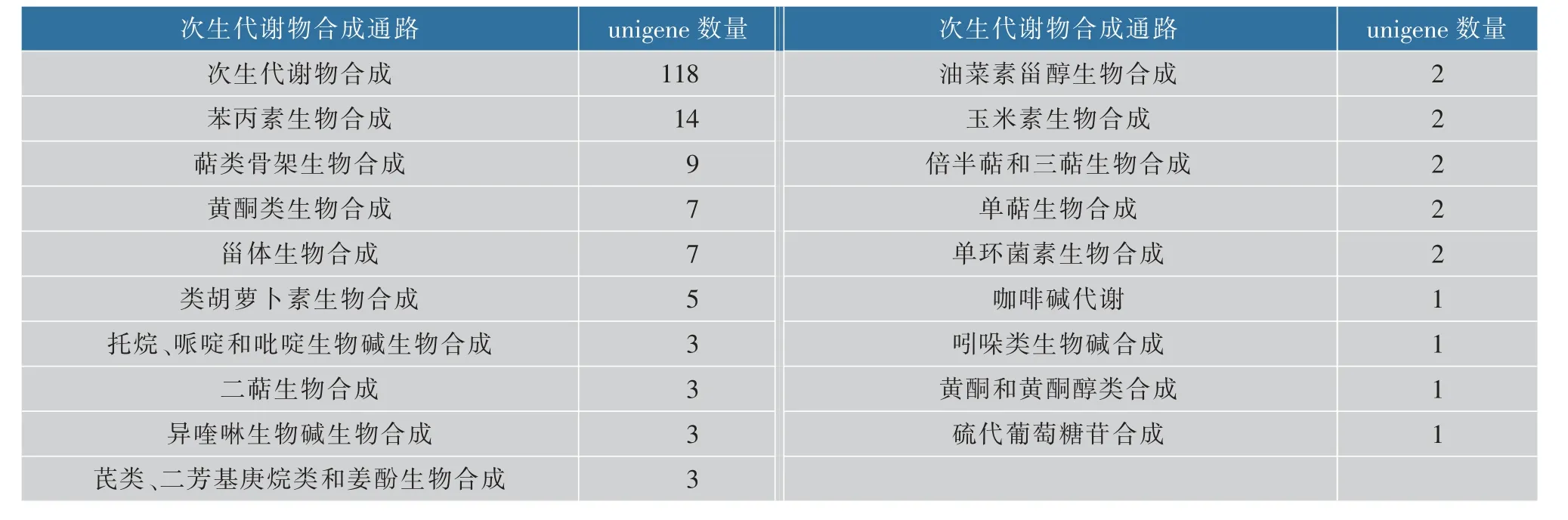
表3 与多花黄精次生代谢相关的unigenesTable 3 Unigenes related to secondary metabolism of P. cyrtonema
2.6 黄酮类化合物合成途径中基因表达差异的初步分析
对多花黄精组培苗在3 个月和9 个月发育期块茎的总黄酮质量分数分析显示,3 个月发育期块茎中总黄酮的平均质量分数为0.008 mg/g,9 个月为0.012 mg/g,表明随着根茎的不断发育,其总黄酮质量分数也在不断积累。 基于FPKM 值比较在3 个月和9 个月发育期块茎的黄酮类化合物合成途径中关键酶基因的表达水平, 发现9 个基因显著上调, 分别是PAL、4CL、DFR、ANS、FLS、UGT、F3′H、COMT 和F3′,5′MT 基因, 上调倍数为1.9~11.2;3个基因显著下调, 分别是CHS、CHI 和F3′,5′H 基因, 下调倍数为0.3~0.5;6 个基因的表达无明显表达差异, 分别是CYP73A、F3H、LAR、CCOMT、HCT和C3′H 基因,这些基因的表达变化倍数为0.9~1.3(见表4)。 这表明随着块茎中总黄酮质量分数的不断积累,其黄酮类化合物合成途径中基因的差异表达并不一致,这也预示了多花黄精复杂的黄酮类化合物合成机制。
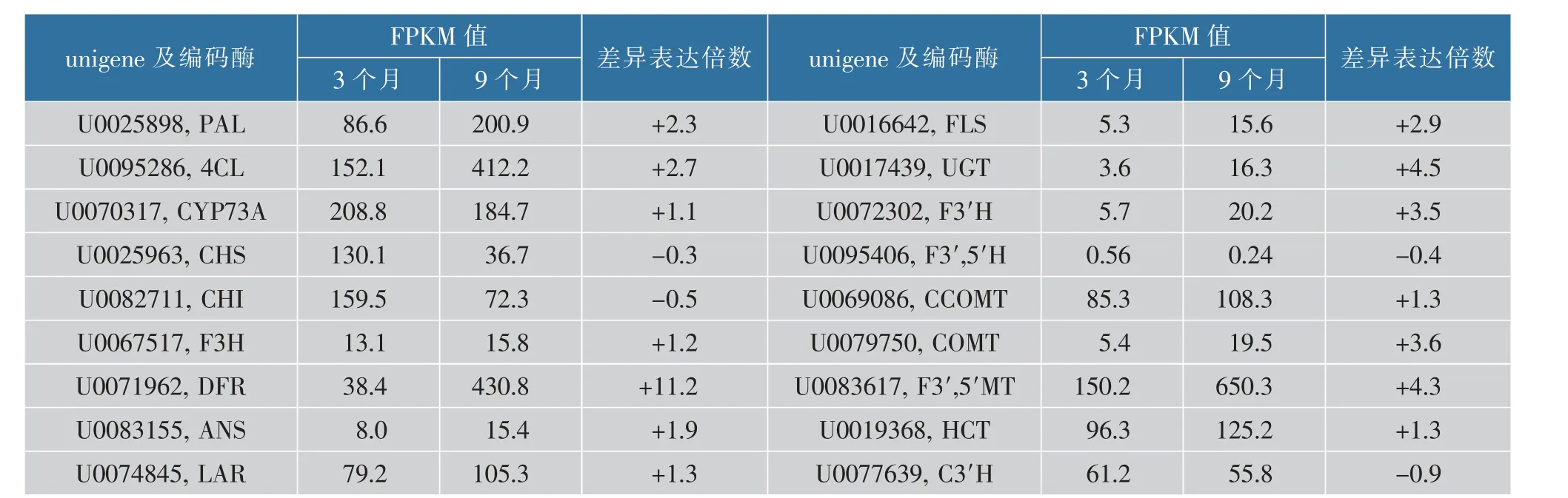
表4 黄酮类化合物合成途径中的基因表达差异Table 4 Different gene expression involved in flavonoids biosynthesis
2.7 RT-qPCR 验证黄酮类化合物生物合成相关酶基因的表达
为验证18 个黄酮类化合物合成酶基因在不同发育期根茎中基于FPKM 值表达变化的可靠性,选取其中的7 个酶基因进一步进行RT-qPCR 分析,并绘制标准曲线。 结果显示,所有酶基因和内参基因(UBQ-E2-10 和EF-1α2)标准曲线的相关系数(R2)均≥0.97,扩增效率均在90%~110%(见表5),表明所有RT-qPCR 引物的扩增具有高特异性, 且扩增效率一致,满足利用2-△△Ct法进行相对定量分析。基因表达结果显示(见图4),这7 个酶基因在不同发育期根茎中的差异表达与转录组FPKM 结果基本吻合,只是这两种分析结果的差异表达倍数略有不同。

表5 用于RT-qPCR 分析的多花黄精黄酮类化合物生物合成相关酶基因Table 5 Genes involved in flavonoid biosynthesis in P. cyrtonema used for RT-qPCR analysis
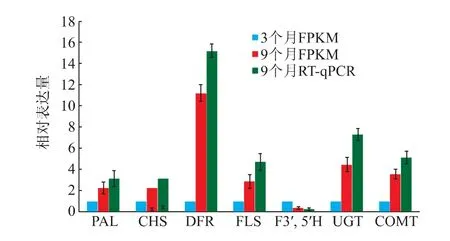
图4 多花黄精7 个黄酮类化合物生物合成相关酶基因的表达Fig. 4 Expression of 7 genes involved in flavonoid biosynthesis in P. cyrtonema
3 结语
利用二代高通量测序技术,对黄精属原生药材多花黄精幼苗期根茎进行转录组测序,经组装拼接共产生了73218 条unigenes 序列, 其中35511 条被注释;共鉴定出83 条unigenes 序列,分别编码18个参与黄酮类化合物生物合成的关键酶;在幼苗期根茎中表达的1359 个转录因子中, 以bHLH 最为丰富,其次是ERF、MYB-relaed、C2H2 和NAC。基于FPKM 表达分析显示, 随着多花黄精块茎中黄酮化合物的不断积累,其黄酮类化合物合成途径中基因的表达并不一致,18 个酶基因中的9 个显著上调,3个显著下调,6 个无明显表达差异。与前期报道的药用植物黄酮类化合物转录组学研究[34-38]相比,本研究鉴定出的黄酮类化合物合成酶的基因数量和种类有些许不同,例如细胞色素CYP450 基因、异黄酮合成酶(IFS)基因等,这可能是由于药用植物在幼苗期根茎发育过程中黄酮类化合物生物合成相关酶基因的表达与叶、花、茎等器官中的表达水平不同而致,也可能与多花黄精的物种特异性有关,这也预示了不同物种间黄酮类化合物的生物合成机理存在一定差异,百合科的黄精属植物可能具有更为复杂的黄酮类化合物生物合成机制。 本研究为后续解析黄精属植物黄酮类活性成分的合成、积累与外源调控机制奠定了基础。
猜你喜欢
食品安全导刊(2021年20期)2021-08-30
中老年保健(2021年11期)2021-08-22
世界科学技术-中医药现代化(2021年10期)2021-03-02
中华养生保健(2020年9期)2021-01-18
Digital Chinese Medicine(2020年3期)2020-11-03
云南农业科技(2020年5期)2020-10-14
河北果树(2020年4期)2020-01-09
现代农业科技(2018年19期)2018-12-21
分析化学(2018年4期)2018-11-02
南方农业·下旬(2017年7期)2017-09-30
