
吸收剂量对二聚酸型聚酰胺结构及性能的影响
2022-04-29 02:08饶小波
辐射研究与辐射工艺学报 2022年2期
饶小波
(上海长园电子材料有限公司 上海 201802)
聚酰胺是指由羧酸类化合物和胺类化合物经缩聚反应生成主链上含有酰胺基的线性热塑性聚合物。聚酰胺分为尼龙型和二聚酸型,其中,二聚酸型聚酰胺是由二聚酸与多元胺经高温缩合反应而成,具有优异的耐化学品性能、耐热性能和耐寒性,快速固化,可粘接多种金属和非金属基材[1‐2],其应用广泛,特别是作为耐高温热熔胶应用于热缩材料领域,作为双壁热缩套管的内胶层,双壁热缩套管受热收缩包覆在基材表面内胶层起到防水密封保护作用[3]。
在热缩材料领域,辐射交联是热收缩产品制造中的重要环节。在制造双壁热缩套管过程中,内胶层材料必然会受到辐射影响,导致分子结构及性能发生变化。以二聚酸型聚酰胺为基材作为双壁热缩套管的内胶层材料,辐射对其分子结构及性能的影响研究较少。
本文通过研究吸收剂量对二聚酸型聚酰胺分子结构及性能的影响,采用傅里叶红外光谱仪研究分子结构的变化,使用差示扫描量热仪(DSC)、软化点测试仪、旋转黏度计及哈克流变仪研究二聚酸型聚酰胺性能的变化,为二聚酸型聚酰胺在热缩材料行业的配方设计开发及应用提供一定的理论依据。
1 材料与方法
1.1 原料与试剂
二聚酸型聚酰胺:软化点(120±5) ℃,Henkel Italia Operations S.r.l.;二甲苯和乙醇均为分析纯,无锡市晶科化工有限公司。
1.2 样品的制备
取一定质量的二聚酸型聚酰胺颗粒,将其放入一定尺寸的平行金属模具中,在温度设定为130 ℃的QLB‐25D/Q 型平板硫化机(常州德杜精密仪器有限公司)上压片,压力设置为0.5 MPa,压片厚度(1.0±0.1)mm,制备的样片待用。
1.3 结构表征及性能测试
1.3.1 辐射交联
常温常压下,在空气氛围中,将压好的样片平铺在平板滑动小车上,在AB0.5‐60 型高频高压型自屏蔽电子加速器(无锡爱邦辐射技术有限公司)上循环辐射,电子束的能量为2.0 MeV,束流强度为32 mA,达到预定吸收剂量(计算见式(1))后停止辐照,制得样品。

式中:D为吸收剂量,kGy;n为辐照次数;I为束流强度,mA;v为平板滑动小车的行进速度,m/min;a为常数,经薄膜剂量计标定取值为24.1。
1.3.2 红外光谱分析
使用美国热电公司(Thermo Electron Scientific Instruments Corp)生产的Nicolet is10 型傅里叶变换红外光谱(FTIR)仪测定,采用ATR‐FTIR,分辨率为6,扫描次数为64,扫描范围650~4 000 cm-1。
1.3.3 差示扫描量热测定
使用美国TA 仪器公司生产的Q20 型差示扫描量热仪测定。N2氛围,以10 ℃/min 的升温速率,从常温开始升温至200 ℃。
1.3.4 软化点测试
采用GB/T 15332—1994 热熔胶胶黏剂软化点的测定方法(环球法)[4],使用上海地质仪器研究所生产的SD‐0606T型软化点测试仪测试。
1.3.5 旋转黏度测试
采用GB/T 2794—2013 胶黏剂黏度的测试方法[5],使用上海昌吉地质仪器有限公司生产的NDJ‐1F型旋转黏度计,选用29号转子测试。
1.3.6 流变学性能分析
采用德国HAAKE(哈克)公司生产的Rheostress1 型流变仪测定。震荡(OSC)变温扫描,温度扫描范围60~180 ℃,频率1.0 Hz,应变1%,采用控制恒定剪切应变式模(CD)。旋转(Rot)定温扫描,温度设定140 ℃,扫描时间300 s,频率1.0 Hz,应变1%,采用控制剪切应力模式(CS)。
1.3.7 凝胶含量测定
依照GB/T 18474—2001 进行凝胶含量的测试[6],将一定质量的二聚酸型聚酰胺样品用不锈钢网包覆,放于二甲苯烧瓶中加热回流12 h,计算回流前后剩余二聚酸型聚酰胺的质量分数,即二聚酸型聚酰胺的凝胶分数(G,%),计算见式(2)。
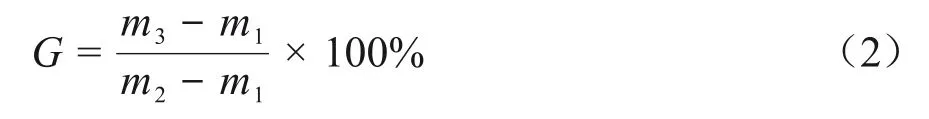
式中:m1为用于包裹二聚酸型聚酰胺试样筛网的质量,g;m2为包裹二聚酸型聚酰胺的筛网总质量,g;m3为经二甲苯回流后,乙醇清洗三次干燥恒重后的总质量,g。
2 结果与讨论
高分子材料在辐射加工过程中,高分子链发生交联反应和降解反应并存[7]。在一定吸收剂量范围,不同种类的高分子材料一般会以其中的一个反应为主[8‐9]。
氧是自由基俘获体,高分子材料在空气中辐照时,主链自由基优先与氧反应生成过氧化物,最终导致主链降解。对于在高剂量率下,如使用电子加速器产生电子束,氧对辐射交联的影响不明显。由于高剂量率下大分子自由基形成的速率快、浓度高、复合交联的概率大;同时高分子材料内的氧消耗快,当另一部分氧还来不及渗入,交联反应已经完成[10‐11]。
在一定吸收剂量范围内,二聚酸型聚酰胺大分子链以交联反应为主[12‐13],反应过程如图1所示。
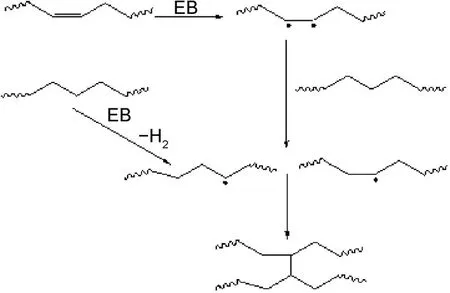
图1 二聚酸型聚酰胺辐射交联反应过程分子结构变化示意图Fig.1 Schematic diagram of molecular structure changes of dimer acid polyamide during radiation crosslinking
二聚酸型聚酰胺大分子链中存在不饱和C=C[14‐15],在辐照过程中双键被激发形成双自由基,继而和大分子链进行夺氢反应形成单自由基,最后两个自由基耦合产生交联[11];同时主链上的-CH2-发生抽氢反应,产生大分子链自由基,大分子链自由基间复合而形成交联[16]。
2.1 FTIR分析
图2为不同吸收剂量的二聚酸型聚酰胺红外谱图。由图2 可知,N-H 的伸缩振动特征峰出现在3 200 cm-1处,3 073 cm-1处为烯氢的伸缩振动特征峰,2 924 cm-1和2 853 cm-1为-CH3和-CH2-的振动特征峰[17], 1 640 cm-1为酰胺基的C=O 伸缩振动峰,l 550 cm-1处的峰是由N-H 弯曲振动和C-N伸缩振动的组合吸收峰,1 253 cm-1的峰也是C-NH 的组合吸收峰,935 cm-1处的峰为烯氢的弯曲振动峰[18‐19],735 cm-1为N-H振动特征峰。随着吸收剂量的增加,红外谱图中酰胺基各特征峰变化不明显,说明在一定的吸收剂量范围内,二聚酸型聚酰胺高分子结构中酰胺基不参与交联反应,由于交联反应主要是C-H 键断裂产生的自由基相互耦合造成的[20]。表征双键的特征峰(3 073 cm-1和935 cm-1)随吸收剂量的增加峰型减弱,说明不饱和键C=C参与交联反应,反应过程如图1所示。

图2 不同吸收剂量的二聚酸型聚酰胺红外谱图:(a)波数:700~3 600 cm-1;(b)波数:700~1 500 cm-1Fig.2 Infrared spectra of dimer acid polyamide with different absorbed doses:(a)wavenumber:700~3 600 cm-1;(b)wavenumber:700~1 500 cm-1
2.2 DSC分析
由图3和表1中的谱图及数据可知,随着吸收剂量的升高,二聚酸型聚酰胺的熔融峰Tm向高温移动,未辐照的二聚酸型聚酰胺Tm为78.0 ℃左右,当吸收剂量为150 kGy 时,Tm升高到85.0 ℃左右。原因是:在辐照过程中,二聚酸型聚酰胺大分子链中C=C形成双自由基,同时主链上的-CH2-发生抽氢反应产生大分子链活性自由基,大分子链自由基间复合而形成交联,大分子链由线性结构变成体型结构(图1),使得大分子链缠绕得更加紧密。在一定吸收剂量范围内,随着吸收剂量增加,大分子链间的交联度升高,导致分子链间的作用力增大,当温度越接近熔点,高分子链段运动越困难,使DSC熔融峰Tm向高温移动[21‐22]。

表1 不同吸收剂量的二聚酸型聚酰胺的熔融峰Tm和熔融焓ΔHmTable 1 DSC melting peak Tm and melting enthalpy ΔHm of dimer acid polyamide at different absorbed doses

图3 不同吸收剂量的二聚酸型聚酰胺DSC谱图Fig.3 DSC spectra of dimer acid polyamide at different absorbed doses
随着吸收剂量增加,熔融焓ΔHm有下降的趋势。说明在本研究的吸收剂量范围内,随着吸收剂量升高,二聚酸型聚酰胺的结晶度有略微下降。一般聚合物有结晶区和无定型区,结晶区内大分子排列规整,分子运动受阻,而无定形区内分子链无规排列,较柔顺,链段活动比较自由。在常温常压下,辐射交联主要发生在无定形区和结晶区表面。若要使结晶区内的大分子链间形成交联,在室温条件下,聚合物必须吸收较高的吸收剂量[11]或者温度接近熔点进行辐照处理[23]。针对二聚酸型聚酰胺结晶区内的辐射交联,尚待深入研究。
2.3 软化点及凝胶含量分析
由图4可知,随着吸收剂量的增加,二聚酸型聚酰胺的软化点和凝胶含量升高明显。未辐照的二聚酸型聚酰胺软化点在123 ℃左右,凝胶含量为0。当吸收剂量为150 kGy 时,软化点升高到158 ℃左右,凝胶含量升高到43%左右。由于未辐照的二聚酸型聚酰胺的大分子链为线性结构,经辐照处理后,分子链间发生交联反应,大分子链转化为体型结构[24]。由凝胶含量数据可知,在一定吸收剂量范围内,随着吸收剂量增加,二聚酸型聚酰胺大分子链间的交联度升高,对应样品的软化点升高,测试结果与§2.2 的DSC 分析结果一致。

图4 不同吸收剂量的二聚酸型聚酰胺软化点和凝胶含量Fig.4 Softening point and gel content of dimer acid polyamide at different absorbed doses
2.4 旋转黏度分析
由图5可知,同一种样品,随着温度升高,旋转黏度的数值降低,由于热熔胶在测试过程中,测试温度越高,大分子链段运动越激烈,分子链间的摩擦阻力降低,旋转黏度计的转子在旋转过程中受到熔体的摩擦阻力越低[25]。在相同的测试温度下,随着吸收剂量增加,二聚酸型聚酰胺的旋转黏度值升高。由于随着吸收剂量升高,二聚酸型聚酰胺的交联度增加,大分子链间通过共价键链接在一起,分子链间的作用力增大,旋转黏度计的转子在旋转过程中受到熔体的摩擦阻力增大,导致旋转黏度数值显著升高,测试结果与§2.3的软化点及凝胶含量的分析结果一致。
图5 不同吸收剂量的二聚酸型聚酰胺黏度Fig.5 Viscosity of dimer acid polyamide with different absorbed doses
2.5 流变学分析
图6为不同吸收剂量的二聚酸型聚酰胺OSC振荡温度扫描谱图分析。由图6及表2可知,储能模量G′和损耗模量G″的交点对应温度Tj随着吸收剂量的增加而升高。当吸收剂量为0 时,Tj为118.2 ℃,当吸收剂量升到120 kGy 时,Tj达到144.7 ℃,而当吸收剂量为150 kGy 时,G′和G″无交点。

表2 不同吸收剂量的二聚酸型聚酰胺的流变学数据Table 2 Rheological data of dimer acid polyamide with different absorbed doses
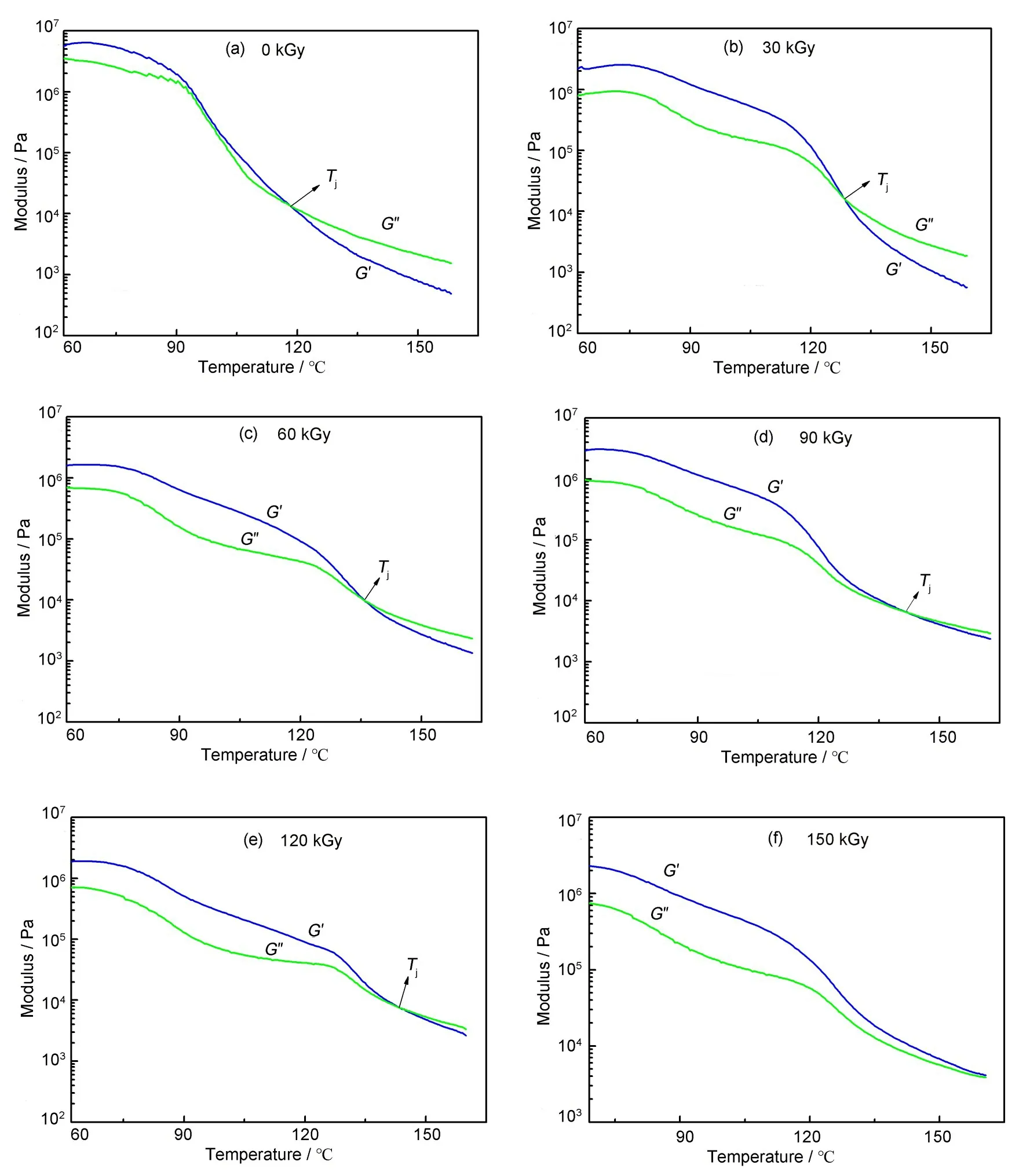
图6 不同吸收剂量的二聚酸型聚酰胺OSC振荡温度扫描谱图Fig.6 OSC oscillating temperature scanning spectra of dimer acid polyamide at different absorbed doses
在G′>G″的温度区间内,高分子材料以弹性为主,在G′<G″的温度区间内,高分子材料以黏性为主。样品在OSC 振荡变温扫描测试条件下,随着温度升高,G′和G″的数值下降,高分子材料由弹性向黏性转变[26]。
损耗模量G″本身的物理意义就是内摩擦的反映,在较低温度下损耗模量过程的综合反映:一是内摩擦使高分子链段取向的过程;二是高分子链段解取向时克服内摩擦的过程,这也使得储能模量高于损耗模量。而在G′和G″的交点处便可以认为是上述高分子链段的内摩擦作用和链段取向作用达到了一种平衡状态[27‐28]。在一定吸收剂量范围内,随着吸收剂量的增加,高分子链间的交联度增加,在相同温度的测试条件下,高分子链段间的内摩擦增大;随着温度升高,高分子链段间的自由体积增大,链段运动变剧烈,高分子链段间的内摩擦减少,使内摩擦作用和链段取向作用的平衡点向高温移动,导致Tj向高温移动。
图7为不同吸收剂量的二聚酸型聚酰胺恒温旋转扫描谱图。由图7 和表2 可知,黏度η随着吸收剂量的增加而增大。黏度η取值为100~300 s 之间的黏度数据,当吸收剂量为0 时,黏度数值为(195±5) Pa·s,当吸收剂量达到150 kGy时,黏度数据达到(2 150±150) Pa·s。原因是:随着吸收剂量升高,高分子链间的交联度增加,高分子链结构由线型结构转化成体型结构,高分子链间的摩擦力增大,该测试结果与§2.4的旋转黏度分析结果一致。

图7 不同吸收剂量的二聚酸型聚酰胺恒温旋转扫描谱图Fig.7 Constant temperature rotating scanning spectra of dimer acid polyamide at different absorbed doses
3 结论
二聚酸型聚酰胺的分子结构及性能受辐射影响显著,红外光谱证明了大分子结构中的C=C 参与辐射交联,性能参数软化点、凝胶含量、旋转黏度、DSC 熔融峰Tm、G′与G″的交点对应温度Tj及黏度η随吸收剂量增大而显著升高。熔融焓ΔHm的数据表明,在本研究的吸收剂量范围内,二聚酸型聚酰胺的结晶度随吸收剂量升高而略微下降。
猜你喜欢
机械工业标准化与质量(2022年8期)2022-10-09
常州大学学报(自然科学版)(2022年2期)2022-04-20
现代仪器与医疗(2022年1期)2022-04-19
现代仪器与医疗(2021年5期)2021-12-02
河北工业大学学报(2020年3期)2020-07-08
当代化工(2019年6期)2019-12-03
中学生数理化·高三版(2016年12期)2017-03-02
电子技术与软件工程(2016年24期)2017-02-23
养生保健指南(2016年9期)2016-05-14
中学生数理化·八年级物理人教版(2016年9期)2016-05-14
