
儿童囊性纤维化1例报道
2022-04-26 06:10:50曹旭阳程燕郭素香
临床肺科杂志 2022年5期
曹旭阳 程燕 郭素香
囊性纤维化(Cystic fibrosis,CF)是一种由于患者机体黏液分泌过多所引起的遗传性外分泌腺疾病,其发病与跨膜传导调节因子(cystic fibrosis trans-membrane conductance regulator,CFTR)基因突变相关,临床主要表现为外分泌腺先天功能紊乱,粘液腺增生,分泌腺粘稠,主要影响胃肠道和呼吸系统。本文主要报道了我院收治的1例儿童囊性纤维化患儿的临床以及病理资料,并通过综述与研究相关的文献作出讨论和总结,旨在进一步提高医学界和临床人员对 CF诊疗的重视度,并提供中国儿童的基因突变谱。
临床资料
患儿,男,10岁,主诉: 间断咳嗽10月余,加重伴间断发热26天。患儿于入院前10月(9岁时)无明显诱因出现咳嗽,阵发性连声咳,有痰,可咳出黄白色粘痰,伴有发热,经住院治疗后,患儿仍有间断咳嗽,入院前26天咳嗽加重,伴有发热,再次住院治疗,根据症状和体征及相关入院常规检查考虑“肺炎、鼻窦炎”,间隔2天后体温正常,咳嗽减轻。入院前4天再次咳嗽加重,阵发性连声咳,有痰,可咳出黄白色粘痰,量中等,伴发热,体温最高达38.5℃,不伴寒战、皮疹、抽搐,口服退热药后可降至正常,间隔24小时体温复升。病程中有流涕、鼻堵,无其他伴随症状。自发病以来,精神可,进食,水可,尿量可,排便2天1次。近10个月患儿体重增加5kg。既往史:患儿生后2月起反复肺炎病史,因肺炎多次住院治疗,平素患儿易感冒,反复流黄涕、咯黄痰;既往鼻炎、中耳炎病史;追溯近年胸部影像学资料及肺功能,提示近3年均为双肺间质性炎性改变,肺功能提示中度限制性通气功能障碍。预防免疫按计划接种。生产史:胎次G1P1,孕周41周,剖腹产,喂养史及生长发育史无异常发现。家族史:一级亲属否认鼻炎、哮喘等类似病史及其他家族遗传性疾病史。体格检查:T 37℃,P 112bpm,RR 28bp,BP 120/80mmHg。神清,精神可,呼吸稍促,节律整齐。鼻通气欠畅,可见黏性分泌物,咽部黏膜充血,扁桃体Ⅱ度肿大。双肺呼吸音粗,双侧肺可闻及细湿啰音及痰鸣音,余未见阳性体征。重要辅助检查:血常规(2020-8-20,本院,编号126)示:WBC 15.44×109/L,N 73.1%, L 21.6%, HGB 127g/L, PLT 472×109/L, CPR 10.18mg/L,肺炎支原体抗体示阴性;胸片(2020-8-20, 本院,X线号D315789)示:双肺纹理增粗,左下肺可见点片状高密度影。尿常规、大便常规、电解质、肝肾功能、心肌酶、CO2cp、GLU未见异常;FER:152.5ng/mL;免疫球蛋白、总IgE均正常范围。呼吸道病原感染九联检:均阴性。肺功能检查:中度限制性通气功能障碍,小气道功能障碍。上气道阻力增高。气道可逆试验:FEV1绝对值提高小于200mL,改善率7.8%,支气管舒张试验后X5下降≥20%。肝脾肾超声提示:肝脾增大;肾脏未见异常。鼻窦CT示:双侧碟窦、筛窦、上颌窦炎症。完善相关检查后予:头孢地嗪抗感染,氨溴索化痰止咳;雾化吸入布地奈德、特布他林、异丙托溴铵、乙酰半胱氨酸溶液加强呼吸管理;口服易坦静止咳祛痰。配合激光行靶向治疗。辅以机械排痰以助痰排出。给药三天后,患儿体温正常,咳嗽减轻,仍咯黄痰,流黄涕,查体左肺可闻及湿性啰音。7天后患儿偶咳,少痰,复查血常规指标正常,好转出院。但是患儿一周后病情再次复发,继查痰培养+药敏:草绿色链球菌。血EB-IgM(+),肺泡灌洗液EB-DNA(+),肺泡灌洗液感染病原体高通量示流感嗜血杆菌序列数。诊断为EB病毒感染,予头孢哌酮舒巴坦钠(约60mg/kg/d)抗感染治疗,雾化加强呼吸道管理及对症支持治疗,两天后体温恢复正常,咳嗽较前减轻,听诊两肺湿性啰音较前吸收。但是该患儿近十年来的反复肺炎病史,因肺炎曾多次住院治疗,继查胸部CT(见图1)以及纤维支气管镜。

图1 胸部CT检查
支气管镜示:支气管内膜炎症(双肺,左肺为著)。
外院呼吸系统遗传病相关基因检测:检测到受检者携带PIK3CD基因一个杂合变异和RPGR基因一个半合子变异,需结合临床情况综合判断。
经患儿监护人同意,对该患儿进行分子遗传检测报告:检测项目:全外显子组检测(三人家系)+CNV。检测结果:检测到受检者2个与临床表型显著相关的变异,以及4个可报告变异(见图2)。
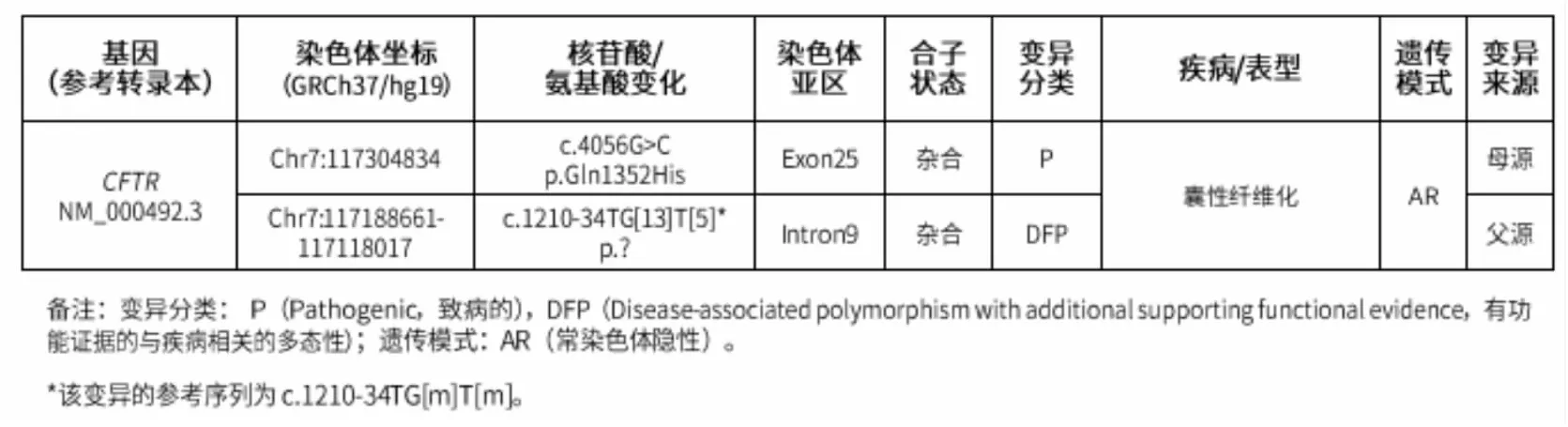
图2 分子遗传检测报告
讨 论
囊性纤维化的发病率在不同人种和所处不同地理环境而表现各异,高加索人受影响最大,高加索人每3000人中就有1人患上该疾病[1],其次是欧美人群,亚洲人发病率相对较少,而针对我国的 CF患者数据报道相对较少,自1974年首次被发现至今,中国人群中 CF共有报道54例[2]。所以目前仍然没有针对中国人群进行的流行病学调查。
囊性纤维化(CF)是一种遗传性外分泌腺疾病,系位于7号染色体长臂3区1带的CFTR基因突变引发。在CF时CFTR的基因编码具有导致其形状异常甚至不在膜内的某些突变,这时氯离子不能离开细胞,失效的CFTR也会导致另一个离子通道的活性增加,被称为上皮钠通道或ENaC,它的机制现仍不详,ENaC允许钠离子进入细胞,最终结果是内部离子浓度比细胞外浓度高,这时渗透会导致粘液中的水穿过细胞膜,进入细胞,使粘液干燥变厚,使纤毛很难移动,不能把粘液扫出气道,在呼吸系统方面会导致感染和气道阻塞;同时在消化道中也发现了CFTR和粘液,刚好在胃和小肠的连接处,这些导管允许胆囊内的胆汁和胰酶进入小肠,这些物质有助于消化食物,失效的CFTR可能再次导致粘液积聚导管中,堵塞导管,阻止这些消化物质的释放,因此CF的另一个症状是消化不良,因为不能正常的分解食物;汗腺中也有CFTR,但是它们允许氯离子进入细胞,由于失效的CFTR,氯离子在汗液中积累并与钠离子结合产生一些盐,因此汗液的异常也是CF的症状之一。
CF的临床表现常为呼吸道症状,如:患儿有慢性、反复的上、下呼吸道感染。咳嗽咳痰,痰液多呈黏稠脓性痰,反复发作的支气管炎丶肺炎甚至肺脓肿以及支扩等,同时大多数 CF 患者还会合并其他疾病,如鼻窦炎等。同时还常常表现有消化道和汗液分泌障碍方面的症状。熊茜萌、徐保平等[3]分析2010年-2017年在中国确诊的26例CF患儿的临床表现及影像学特征,结论如下:反复咳嗽咳痰等呼吸系统症状发病率为100%,存在鼻窦炎病史占76.9%,营养不良的发病率为34.6%,腹泻腹胀11.5%,生长迟缓3.8%,胎粪肠梗阻发病率为0。
本例患儿在出生后2个月起,即有反复的肺炎病史,发病时咳嗽、咯大量黄痰,鼻塞、流黄涕,既往有鼻窦炎、中耳炎病史,但是无家族阳性病史,营养状况和生长发育情况尚可。
诊断标准[4]:必须同时满足以下两条标准,才能确定其是CF: 至少1个器官系统的临床症状符合CF; 存在CFTR 功能障碍证据(以下任1条):(1)出现汗液中氯化物水平上升至≥60 mmol/L(每次检测2次);(2)CFTR 存在2个致病性突变,亲代等位基因各自提供1个;由于各个种族、所在地区、所处的特殊地理环境不同, CFTR 常见的位置和突变不同[5-6]。所以对中国疑似CF患者进行CFTR基因全外显子测序,不仅能够找到少见的基因突变甚至新基因突变。
肺部病变的治疗:其目的主要是为了清除呼吸气道内的分泌物以及控制感染的慢性传播[7]。静脉应用的抗生素,主要分为β内酰胺类及氨基糖苷类抗生素,通常主要应用于严重感染的患者[8-9]。肺部疾病情况严重时,可以考虑进行肺移植[10]。消化道病变治疗和营养支持治疗:给予高热量、高蛋白饮食。并且要注意补充适量食盐。Corey 等[11]的一些研究结果表明,CF患儿中,营养状况越差,肺部细胞感染发生几率就会越高,预后更差。基因治疗:2012年获得FDA批准上市的基因治疗药物有治疗G551D突变的Ivacaftor(Kalydeco)。据临床医学专家报道,患者初次应用2周后的临床病情症状即可明显地逐渐得到基本好转,效果最长时间可以持续保持48周,肺功能、肺部病变急性加重次数及汗液氯离子浓度等多项指标,均明显得到改善[12]。2019年10月,美国FDA批准Trikafta(elexacaftor/ivacaftor/tezacaftor)目前最新提出来治疗囊性纤维化突变患者的三联疗法,主要用于治疗12岁及以上,且至少携带一个F508del基因突变的CF患者(约占CF患者的90%),这一疗法成为目前很大一部分CF患者群体的治疗药物[13]。
总 结
由于CF患儿的早期发病在目前我国相对较为少数,罕见,且其临床表现不典型,容易错过最早的诊断和治疗时机,所以针对该病的早发现、早诊断、早治疗尤为重要。(1)本例患儿自出生2个月开始有反复的肺炎病史,间断咳嗽时间长,时有发热,咳嗽伴痰量较多,总咯黄痰,吼吼的痰声,肺部听诊可闻及湿啰音以及痰鸣音,符合儿童湿性咳嗽的范畴。(2)进一步诊断要鉴别是因为感染、哮喘等引起的粘液高分泌,还是因为原发性纤毛运动障碍及呼吸道感染(病毒性、细菌性等)、空气污染等引起纤毛运动障碍,或者是自身免疫性疾病、囊性纤维化、支气管扩张、气道畸形、气道异物等引起的黏液清除障碍。(3)本例患儿住院期间经过西医的抗感染以及对症治疗,给药三天后,患儿体温正常,咳嗽减轻,仍咯黄痰,流黄涕,查体左肺可闻及湿性啰音。经中西医结合治疗7天后患儿偶咳,少痰,复查血常规指标正常,好转出院。但是出院后1个月又再次出现肺部相关症状。(4)此时胸部高分辨CT、纤维支气管镜、基因检测虽然价格昂贵,但是在临床中的诊断和排除诊断具有很高的临床价值。
猜你喜欢
中国毕业后医学教育(2021年2期)2021-12-06 06:09:20
世界最新医学信息文摘(2020年51期)2020-12-25 17:47:25
中国毕业后医学教育(2020年6期)2020-12-06 07:30:02
智能计算机与应用(2019年4期)2019-09-12 10:41:42
中国临床医学影像杂志(2019年1期)2019-04-25 06:49:38
心肺血管病杂志(2019年1期)2019-04-22 01:12:02
中国卫生(2016年9期)2016-11-12 13:28:06
海南医学(2016年8期)2016-06-08 05:43:00
长江大学学报(自科版)(2014年12期)2014-03-20 13:21:41
长江大学学报(自科版)(2013年33期)2013-03-11 15:08:19
