
致病性Th17细胞在神经炎症中的作用及调控机制的研究进展
2022-04-24 05:18戴鸿宇季东谈程孙杰姚昊
遗传 2022年4期
戴鸿宇,季东,谈程,孙杰,姚昊
致病性Th17细胞在神经炎症中的作用及调控机制的研究进展
戴鸿宇1,2,季东1,2,谈程1,2,孙杰3,姚昊1,2
1. 南京医科大学第二附属医院心血管中心,南京 210003 2. 南京医科大学第二附属医院麻醉科,南京 210011 3. 东南大学附属中大医院麻醉科,南京 210009
神经炎症是中枢神经系统在损伤、感染、毒素等各种影响内稳态因素的刺激下产生的复杂免疫反应,涉及驻留在中枢神经系统中的多种免疫细胞。持续存在的神经炎症是所有神经系统疾病(包括神经发育、神经退行性和精神性疾病)病因和病程的共同特性。Th17细胞是CD4+T细胞的一个重要亚型,在稳态条件下介导对细胞外细菌和真菌的免疫反应,维持肠道粘膜屏障的防御功能。但当体内细胞因子微环境发生炎症性改变时,Th17细胞可以转化为具有高度促炎性的致病表型,在炎症性疾病的发生发展中起着至关重要的作用。本文主要对致病性Th17细胞的分化调控及其在神经炎症中的作用进行了系统综述,对于理解免疫系统和神经系统之间的相互作用具有一定参考意义。
神经炎症;致病性Th17细胞;血脑屏障;RORγt
炎症是机体对损伤、感染或其他刺激做出的防御性反应,一方面,它能够消除致病因素并修复组织损伤,另一方面,当炎症反应过激或趋于慢性时,会形成炎症环境,免疫系统转而攻击自身组织和细胞,导致进行性损伤。神经炎症是中枢神经系统(central nervous system, CNS)在损伤、感染、毒素等各种影响内稳态因素的刺激下产生的复杂免疫反应,涉及CNS中的免疫细胞和多种驻留细胞,包括胶质细胞(小胶质细胞、星形胶质细胞、少突胶质细胞)、髓样细胞(巨噬细胞和树突状细胞)以及外周白细胞。持续存在的神经炎症是神经系统疾病(如神经发育、神经退行性和精神性疾病)病因和病程的共同特性[1]。许多研究表明,神经炎症的严重程度与Th17细胞介导的免疫反应之间存在相关性[2]。Th17细胞作为已证实的致病细胞与多发性硬化症(multiple sclerosis, MS)[3]、帕金森病(Parkinson’s disease, PD)[4]等多种神经炎症参与的中枢神经系统退行性疾病息息相关。本文对致病性Th17细胞的分化调控及其在神经炎症中的作用进行了系统综述,深入阐述了致病性Th17细胞的产生和致病性的分子调控机制,对于理解免疫系统和神经系统之间的相互作用、预防中枢神经系统疾病并延缓其进展具有一定参考意义。
1 Th17细胞概况
Th17细胞是一类独立于Th1和Th2的CD4+T细胞亚型,产生标志性细胞因子IL-17,特异性表达维甲酸相关孤儿受体γt (retinoic acid-associated orphan receptor gamma t, RORγt)和信号转导与转录激活因子3 (signal transducer and activator of transcription 3, STAT3)两种转录因子,自2005年发现以来Th17细胞引起了人们的极大关注[5]。在稳态条件下,Th17细胞介导对细胞外细菌和真菌的免疫反应,维持肠道粘膜屏障的防御功能[6],但当体内细胞因子微环境发生炎症性改变时,Th17细胞可以转化为具有高度促炎性的致病表型,突破血脑屏障(blood- brain barrier, BBB)并招募更多的免疫细胞参与神经炎症,导致神经变性。鉴于Th17细胞在炎症性疾病和自身免疫性疾病的发生发展中具有重要作用,目前已被用于临床免疫治疗的靶标。Th17细胞对宿主既有致病性,又有非致病性,而分化为哪一种表型则取决于不同的细胞因子微环境[7]。鉴于此,越来越多的研究致力于揭示Th17细胞的异质性及其调控机制。
1.1 Th17细胞的异质性
Th17细胞的表型和功能特性在体外和体内已经得到了广泛的研究。众多研究结果表明,Th17细胞具有异质性,体现为既有免疫抑制调节性又有高度促炎性[8]。根据过继转移后是否具有诱导实验性自身免疫性脑脊髓膜炎(experimental autoimmune encephalomyelitis, EAE)的能力,可将Th17细胞分为致病性Th17细胞(pathogenic Th17 cells, pTh17 cells)和非致病性Th17细胞(non-pathogenic Th17 cells, non-pTh17 cells)两种表型[9]。
1.2 非致病性Th17细胞的产生与功能
转化生长因子β1 (transforming growth factor beta 1, TGF-β1)和IL-6诱导的Th17细胞多数情况下被认为是non-pTh17细胞(图1),甚至在某些肠道疾病中表现出保护作用[10,11]。它表达免疫调节细胞因子IL10和IL-4,以及CD5抗原样蛋白(CD5 molecule- like, CD5L)、IL-9和GATA3 (GATA binding protein 3)等一系列病理生理反应的负性调节因子[12]。在体内稳定状态下,分枝丝状杆菌(segmented filamentous bacteria, SFB)诱导生成的non-pTh17细胞驻留在小肠固有层并分泌大量IL-10,具有维持肠道的免疫平衡和组织完整性的功能[6],这种免疫调节和组织驻留功能依赖于c-Maf的调控[13]。CD5L属于富含半胱氨酸的清道夫受体超家族,参与脂质代谢的调节,特别是在维持多不饱和脂肪酸/脂肪酸水平的平衡中发挥作用。CD5L在non-pTh17细胞中特异性上调,通过限制其胆固醇衍生配体的获取来抑制RORγt的转录活性[9]。
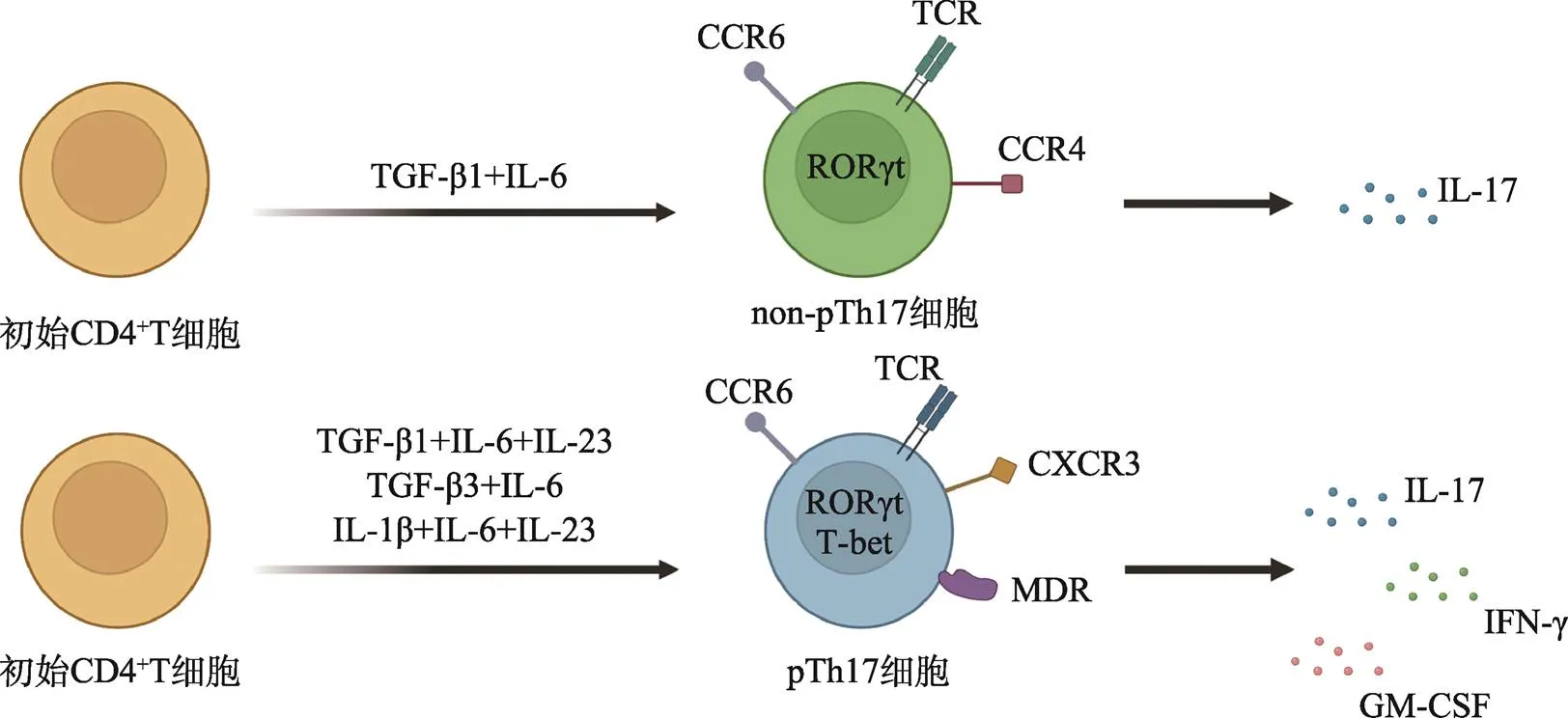
图1 Th17细胞分化途径、信号转导因子及细胞因子示意图
TGF-β1联合IL-6有助于Th17细胞的极化,而IL-1β、IL-6、IL-23、TGF-β3的加入使Th17细胞进一步分化为致病性Th17细胞,参与神经炎症性疾病的发生发展。根据参考文献[9],使用BioRender.com修改绘制。
1.3 致病性Th17细胞的产生与功能
在抗原呈递细胞(antigen presenting cells, APCs)分泌的IL-23的刺激下,non-pTh17将转化为具有致病作用的pTh17细胞[14],这表明IL-23/IL-23R通路在pTh17细胞诱导炎症反应中起着重要作用,缺乏IL-23R (IL-23 receptor)会降低pTh17细胞的致病潜能,并减少其在CNS的聚集。pTh17细胞在CNS中的致病性一方面有赖于其高度表达的粒细胞巨噬细胞集落刺激因子(granulocyte-macrophage colony- stimulating factor, GM-CSF),这主要由IL-23和RORγt介导[15],而GM-CSF又可以通过作用于树突状细胞(dendritic cells, DCs)提高pTh17细胞IL-23的产量[16],从而形成一个正反馈回路。另一方面,pTh17细胞兼有Th17和Th1细胞的促炎特性和致病特征,可同时分泌IL-17和IFN-γ (Th1的标志性细胞因子),且共同表达转录因子RORγt/RORC [小鼠()/人类()]和T-bet (T-box expressed in T cells,Th1细胞的特异性转录因子),因此亦被命名为Th17.1细胞[17]。
与non-p17细胞相比,具有强烈Th1倾向性的pTh17细胞具有更强的破坏BBB的能力,使炎症细胞更易于向CNS浸润[18]。激活后的pTh17细胞具有更高的增殖效率,并诱导多药耐药蛋白1 (multidrug resistance protein 1, MDR1)的表达,而MDR1能潜在地保护pTh17细胞免受治疗药物的影响[19]。此外,pTh17细胞对Tregs (regulatory T cells)的抑制可能有一定的抵抗力[20]。总的来说,pTh17细胞具有一些典型的炎症细胞特征,这使它们在CNS中具有高度致病性(图1)。
2 致病性Th17在神经炎症中的作用机制
pTh17细胞参与神经炎症的发生发展已在EAE和MS的相关研究中证实,而pTh17细胞衍生的细胞因子亦可直接或间接地增强中枢神经系统的炎症反应[21]。pTh17细胞诱导并参与神经炎症的相关机制主要包括如下方面(图2)。

图2 致病性Th17细胞参与神经炎症的分子机制示意图
外周血中的pTh17细胞通过产生促炎细胞因子和细胞黏附分子相互作用突破ECs,削弱BBB的屏障作用,使大量免疫细胞和促炎介质涌入CNS,进一步破坏BBB。在CNS中,pTh17细胞的浸润激活小胶质细胞,IL-17/IL-17R信号决定其炎症因子的表达,并增加ROS的产生,促进中性粒细胞的趋化和聚集。活化的小胶质细胞启动神经元凋亡途径,最终导致神经变性。根据参考文献[27,30],使用BioRender.com修改绘制。
2.1 破坏血脑屏障
BBB是大脑和血液之间的一层内皮屏障,能够选择性地阻止某些物质和免疫细胞进入脑实质,并为CNS提供氧气和关键营养物质。高度特化的内皮细胞(endothelial cells, ECs)通过与周细胞、血管周围星形胶质细胞和神经元相互作用,形成神经血管单元(neurovascular unit, NVU),限制细胞和溶质的细胞旁和跨细胞运动,确保了BBB的功能性和完整性[22,23]。穿越BBB是pTh17细胞参与神经炎症的第一步。体外和体内研究表明,分泌IFN-γ的pTh17细胞能够优先穿越人类的BBB,且在EAE小鼠和MS患者的脑实质病变区积累,表现出神经毒性作用[24]。这一特性依赖于pTh17细胞表达的特殊粘附分子及其分泌的细胞因子。
pTh17细胞能够以MCAM/MCAM的方式粘附ECs,特异性阻断MCAM (melanoma cell adhesion molecule)可以限制pTh17细胞的迁移行为并降低EAE的严重程度[25]。趋化因子受体6 (chemokine receptor 6, CCR6)是RORγt/RORc诱导的一种Th17细胞特征性表面分子,其配体CCL20在炎症部位如脉络丛上皮细胞密集表达,两者之间的相互作用亦参与了pTh17细胞在CNS的浸润过程[26]。其他的表面粘附分子相互作用对,如ICAM-1/LFA-1 (intercellular adhesion molecule-1/lymphocyte function- associated antigen-1)、VCAM-1/VLA-4 (vascular cell adhesion molecule-1/very late antigen-4)、E-selectin/ ESL-1 (E-selectin ligand-1)、ICAM-1/Mac-1 (macrophage-1 antigen)以及P-selectin/PSGL-1 (P-selectin glycoprotein ligand-1)等,在pTh17细胞粘附BBB紧密连接区域的ECs方面发挥作用[27]。
pTh17细胞标志性表达的IL-17是其进一步破坏BBB的主要效应因子。IL-17可促进IL-6、MIP-2 (macrophage inflammatory protein-2)、NO和黏附分子的产生,促进中性粒细胞浸润[28]。中性粒细胞分泌基质金属蛋白酶(matrix metalloproteinases, MMPs)、明胶酶和蛋白酶等各种酶类,使BBB的屏障功能进一步瓦解,有助于免疫细胞的顺利突破。此外,IL-17增加了CNS中活性氧(reactive oxygen species, ROS)的产生,ROS增多导致ECs黏附分子上调,促进单核/巨噬细胞等其他炎性细胞向脑实质迁移[29]。
2.2 激活胶质细胞
增强小胶质细胞功能是与IL-17相关的另一个致病特征[30]。浸润脑实质后,pTh17细胞通过IL-17/IL-17R信号激活小胶质细胞,增加其表面共刺激分子(如ICAM-1、CD40、CD80和CD86等)和MHC-II的表达。活化的小胶质细胞分泌IL-1β、TNF-α、IL-6、补体蛋白和ROS等,在神经炎症中起关键作用[31],其中IL-1β、IL-6和TNF-α (tumor necrosis factor alpha)可启动神经元凋亡通路,最终导致神经退行性病变。
血管周围星形胶质细胞是BBB重要的结构和功能组成部分之一,其末端与内皮细胞层相互作用,包绕大脑的脉管系统[32]。星形胶质细胞表达IL-17R (IL-17 receptor),IL-17与之结合后会刺激星形胶质细胞向反应型表型极化,并产生多种炎症因子和趋化因子[33,34]。IL-17还促进星形胶质细胞表达CCL20[33,35],从而加速pTh17细胞向CNS迁移。此外,Th17细胞相关细胞因子上调脑干星形胶质细胞表面VCAM-1的表达,进一步促进CNS内炎症细胞的聚集[36]。
2.3 损伤神经元
在神经炎症背景下,pTh17细胞触发IL-1β、IL-6和TNF-α等一系列促炎细胞因子的产生,它们与神经元表面的受体结合介导神经元凋亡[37]。靶向谷氨酸兴奋性毒性是pTh17细胞损伤神经元的另一效应途径,主要指向受VCAM-1/integrin β1/KV 1.3信号轴控制的神经元[38]。此外,pTh17细胞可以通过细胞间直接接触的机制诱导神经元内钙离子升高,导致轴突肿胀和细胞死亡,亦可通过Fas/FasL相互作用直接诱导神经元凋亡[39]。
3 致病性Th17细胞分化的调控机制
3.1 IL-23
初始CD4+T细胞缺乏IL-23R,因此IL-23并不参与Th17细胞的极化[40],但在稳定和增强Th17细胞表型方面,IL-23具有关键作用。通过诱导IL-23R的表达,IL-23赋予了Th17细胞致病效应,通过STAT3机制稳定Th17细胞的致病表型。Hirota等[41]的研究表明,pTh17细胞上T-bet和IFN-γ的表达也依赖于IL-23的存在。IL-23/IL-23R信号共同促进Th17细胞的稳定和存活,这对于Th17细胞获得致病特性是必不可少的。IL-23抑制CD5L的表达并调节Th17细胞的代谢状态,在IL-23下游的转录因子Blimp-1 (B-lymphocyte-induced maturation protein-1)亦可驱动pTh17细胞的致病程序,同时抑制pTh17细胞的抑制因子IL-2和Bcl6 (B-cell lymphoma 6)[42]。
血清糖皮质激素激酶1 (serum glucocorticoid kinase 1, SGK1)是一种丝氨酸/苏氨酸激酶,是IL-23信号传导的一个重要节点。体外实验表明,适当增加盐浓度诱导SGK1表达,促进IL-23R表达并增强pTh17细胞分化,从而加速自身免疫疾病的发展[43]。对EAE动物模型的研究进一步表明,高盐饮食会加剧疾病严重程度,并伴随pTh17细胞在脊髓中的浸润增加[44]。这些数据表明SGK1在调节IL-23R表达和维持pTh17细胞表型方面具有关键作用,提供了环境因素(如高盐饮食)触发pTh17细胞发育并促进组织炎症的分子机制。
3.2 GM-CSF
在神经炎症中,另一个介导Th17细胞致病性的重要细胞因子是GM-CSF。GM-CSF是一种与自身炎症相关的促炎症细胞因子,它可以促进DCs的成熟以及粒细胞和巨噬细胞的活化,并使髓样细胞从骨髓动员到外周[45]。pTh17细胞中GM-CSF的增加涉及以下两种机制:(1) IL-23和RORγt驱动Th17细胞中GM-CSF的产生[15];(2) GM-CSF作用于DCs以增强其IL-23的表达,进而促进pTh17细胞的进一步激活和GM-CSF的产生[16]。El-Behi等[46]指出,在中枢神经系统自身免疫的背景下,IL-1β和IL-23诱导Th17细胞产生GM-CSF,并且GM-CSF在脑致病性中具有重要作用;缺乏GM-CSF的pTh17细胞,尽管可以产生IL-17和IFN-γ,却不能导致神经炎症性疾病。
3.3 TGF-β超家族
TGF-β是TGF-β超家族成员之一,包括TGF-β1、TGF-β2和TGF-β3三种亚型[47],TGF-β相关信号通路在体内和体外对Th17细胞的分化起重要作用[48~50]。T细胞是TGF-β1的重要来源之一,极化后的Th1、Th2和Th17细胞都可以表达TGF-β1,但表达量增加最为明显的是Th17细胞[51]。初始T细胞分化为Th17细胞有赖于TGF-β1的自分泌[49],但TGF-β1并不会影响IL-17A的表达,因为从f/fCD4- Cre+小鼠(T细胞中TGFβRI信号被阻断的小鼠)的肠道中仍可检测到产生IL-17A的CD4+T细胞[52]。TGF-β不仅促进Th17细胞分化,还参与决定Th17细胞的致病性。与TGF-β1相比,TGF-β3联合IL-6诱导的Th17细胞在EAE中具有更强的致病作用[7,52],且TRIM28 (tripartite motif-containing 28)缺陷引起的TGF-β3过表达会大大促进pTh17细胞的发育和累积[53]。在TGF-β信号转导通路中,TGF-β超家族成员作为配体与受体结合后会激活不同的Smad蛋白亚型,从而调控不同靶基因的特异性表达,这对TGF-β信号转导的最终生物学效应起着决定性作用[54]。虽然TGFβ3与TGFβ1都与TGFβRII (ALK5)结合,但TGFβ3在pTh17细胞中诱导了Smad1/5的激活,而不是经典的Smad2/3信号。这些研究表明,TGFβ3诱导的pTh17细胞从发育途径上就不同于non-pTh17细胞。
激活素A (activin-A)是也TGF-β超家族的成员之一,是一种与TGF-β1密切相关的多效性细胞因子,可以调节组织稳态、细胞增殖和组织炎症[55]。在体外,activin-A可以诱导Th17细胞分化[56],这一点与TGF-β1类似。但最近的一项研究发现activin-A和TGF-β1对pTh17细胞的作用截然不同。TGF-β1与ALK5的结合抑制ERK (extracellular signal-regulated kinase)磷酸化,而activin-A与其受体(AKL4)结合后激活ERK[57],ERK磷酸化激活赋予了Th17细胞致病能力[58,59],因此,内源性activin-A/ALK4/ERK通路对于促进pTh17细胞介导的神经炎症的至关重要。作为调节TGF-β/activin-A/Nodal信号传导的因子,磷酸酶PP2A (protein phosphatase 2A)通过调控Smad2和Smad3的磷酸化来促进pTh17细胞的产生,并介导EAE效应[60]。这些发现表明,TGF-β超家族通过多种复杂的分子网络在pTh17细胞的产生和功能中发挥着广泛的作用。
3.4 MicroRNAs
MicroRNAs (miRNAs)是单链、约22 nt的非编码RNA,是复杂的基因表达网络在转录后水平上的关键调控因子,参与包括细胞发育和分化在内的多种生物过程。在271个物种中大约有38,589个miRNA前体,产生48,860个成熟的miRNA[61]。对MS患者T细胞中miRNA图谱的分析以及众多基于EAE动物模型的研究表明,miRNAs可能在调控Th17细胞分化和MS病理生理过程中发挥重要作用[62,63]。
miR-183C包含miR-183、miR-96和miR-182三种miRNA,受Dicer1调控,在pTh17细胞中显著表达,通过抑制FOXO1 (forkhead box protein O1)促进Th17细胞产生致病特性[64]。在Th17极化的初始T细胞中,IL-6通过IL-6/STAT3信号上调miR-183C表达,其中miR-96可特异性地促进pTh17细胞产生IL-17A、IL-17F、IL-22和GM-CSF等炎症因子,从而放大pTh17细胞的致病效应,使EAE动物的病理评分明显升高,而TGF-β则下调miR-183C的表达。miR-448是一种在肿瘤细胞中异常表达的miRNA,广泛参与增殖、凋亡、侵袭和上皮-间充质转化等病理生理过程[65~68]。最近有研究者在MS患者脑脊液和PBMCs (peripheral blood mononuclear cells)中检测到miR-448的异常表达,且其表达水平与疾病严重程度正相关。进一步研究EAE小鼠发现miR-448可上调IL-17A与RORγt的表达,促进pTh17细胞分化,亦可直接靶向PTPN2 (protein tyrosine phosphatase non-receptor type 2),抑制其对Th17细胞分化的抑制作用[69]。与健康对照者相比,miR-20b是MS患者PBMCs中唯一下调的miRNA[70],使用慢病毒载体在体内过表达miRNA-20b可减少pTh17细胞数量,降低EAE严重程度,其下游靶点可能是RORγt和STAT3[71]。
3.5 其他调节靶点
RNA结合蛋白HuR是胚胎致死异常视觉(embryonic lethal abnormal vision, ELAV)家族的成员,敲除HuR可降低转录因子RORγt、IRF4 (interferon regulatory factor 4)、RUNX1 (runt-related transcription factor 1)和T-bet的水平,从而减少EAE模型中IL-17+IFN-γ+CD4+T细胞的数量[72]。另一项研究表明,HuR通过结合和稳定CCR6 mRNA以及促进翻译来调节CCR6的表达,敲除HuR降低了Th17细胞表面的CCR6表达水平,抑制其向CNS的迁移能力,从而改善EAE[73]。这些发现凸显了HuR促进Th17细胞介导的自身免疫性神经炎症的分子机制,提示HuR是治疗CNS自身免疫性疾病的潜在靶标。
Roy等[74]发现活化的T细胞使用外源性蛋氨酸合成S-腺苷蛋氨酸(S-adenosyl methionine, SAM),而限制蛋氨酸摄入可降低T细胞内SAM和组蛋白H3K4me3 (histone 3 lysine 4 trimethylation)水平,并通过限制pTh17细胞的扩增来改善EAE的发作和严重程度;该研究证明了限制蛋氨酸饮食能够通过影响Th17细胞的增殖及其细胞因子的产生来影响T细胞介导的自身免疫。Jonathan等[75]提出E3泛素连接酶Hectd3 (HECT domain-containing E3 ubiquitin ligase 3)对MALT1 (mucosa-associated lymphoid tissue lymphoma translocation 1)和STAT3的非降解性泛素化修饰,分别导致NF-κB (Nuclear factor-κB)活化和RORγt上调,促进EAE中pTh17细胞分化,且–/–小鼠的EAE症状改善与IL-17A和GM-CSF表达降低、RORγt下调以及–/–CD4+T细胞中STAT3 Y705位点磷酸化的减少相关,揭示了泛素化影响Th17细胞分化和功能的具体机制。
4 结语与展望
从2005年首次发现至今,对于Th17细胞分化、功能和调控的探索已经取得了飞跃式的进展。随着技术的不断革新,对细胞异质性的研究手段也从流式细胞技术[76]和基于细胞群体的基因组图谱分析进入单细胞测序(single cell RNA sequencing)时代[77]。Gaublomme等[78]在疾病高峰期分离EAE小鼠CNS和引流淋巴结中的Th17细胞,随后进行单细胞测序,并与体外分化的pTh17细胞和non-pTh17细胞测序结果进行比对,结果显示Th17细胞的致病性不仅是由于促炎基因()表达上调,也与免疫抑制基因()的下调有关。这些与Th17细胞异质性相关的基因既有已知的调节因子,也包括了新的候选基因,且两种细胞的基因组图谱的有一定程度的重叠。研究者利用基因敲除小鼠进一步验证后,筛选出了和四个极具潜力的候选基因[12,76],希望能够在不影响non-Th17细胞及机体屏障功能的前提下,约束pTh17细胞的致病能力,延缓神经炎症的发生发展。目前仍有许多未知的领域亟待进一步的科学探索,如pTh17和non-pTh17细胞之间的产生及分化的具体分子机制、如何在病理条件下更明确地区分二者以及能否逆转pTh17细胞致病性等,而这些谜题的揭秘将有助于未来找寻出更多Th17相关神经炎症疾病的精准治疗方法。
[1] Solleiro-Villavicencio H, Rivas-Arancibia S. Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4+T cells in neurodegenerative diseases., 2018, 12: 114.
[2] Singh RP, Hasan S, Sharma S, Nagra S, Yamaguchi DT, Wong DTW, Hahn BH, Hossain A. Th17 cells in inflammation and autoimmunity., 2014, 13(12): 1174–1181.
[3] Moser T, Akgün K, Proschmann U, Sellner J, Ziemssen T. The role of TH17 cells in multiple sclerosis: therapeutic implications., 2020, 19(10): 102647.
[4] MacMahon Copas AN, McComish SF, Fletcher JM, Caldwell MA. The pathogenesis of Parkinson’s disease: a complex interplay between astrocytes, microglia, and T lymphocytes?, 2021, 12: 666737.
[5] Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages., 2005, 6(11): 1123–1132.
[6] Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, Li Y, Kelly G, Chakravarty P, Shoaie S, Gutierrez MG, Stockinger B. The intestine harbors functionally distinct homeostatic tissue-resident and inflammatory Th17 cells., 2019, 51(1): 77–89.e6.
[7] Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells., 2012, 13(10): 991–999.
[8] Ghoreschi K, Laurence A, Yang XP, Hirahara K, O’Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease., 2011, 32(9): 395–401.
[9] Stockinger B, Omenetti S. The dichotomous nature of T helper 17 cells., 2017, 17(9): 535–544.
[10] Dankers W, Davelaar N, van Hamburg JP, van de Peppel J, Colin EM, Lubberts E. Human memory Th17 cell populations change into anti-inflammatory cells with regulatory capacity upon exposure to active vitamin D., 2019, 10: 1504.
[11] O'Connor W, Kamanaka M, Booth CJ, Town T, Nakae S, Iwakura Y, Kolls JK, Flavell RA. A protective function for interleukin 17A in T cell-mediated intestinal inflammation., 2009, 10(6): 603–609.
[12] Wang C, Yosef N, Gaublomme J, Wu C, Lee Y, Clish CB, Kaminski J, Xiao S, Meyer Zu Horste G, Pawlak M, Kishi Y, Joller N, Karwacz K, Zhu C, Ordovas-Montanes M, Madi A, Wortman I, Miyazaki T, Sobel RA, Park H, Regev A, Kuchroo VK. CD5L/AIM regulates lipid biosynthesis and restrains Th17 cell pathogenicity., 2015, 163(6): 1413–1427.
[13] Aschenbrenner D, Foglierini M, Jarrossay D, Hu D, Weiner HL, Kuchroo VK, Lanzavecchia A, Notarbartolo S, Sallusto F. An immunoregulatory and tissue-residency program modulated by c-MAF in human TH17 cells., 2018, 19(10): 1126–1136.
[14] McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology., 2007, 8(12): 1390–1397.
[15] Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation., 2011, 12(6): 560–567.
[16] Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival., 2008, 205(10): 2281–2294.
[17] Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-γ- producing Th17 cells is independent of T-bet., 2013, 190(9): 4478–4482.
[18] van Langelaar J, van der Vuurst de Vries RM, Janssen M, Wierenga-Wolf AF, Spilt IM, Siepman TA, Dankers W, Verjans GMGM, de Vries HE, Lubberts E, Hintzen RQ, van Luijn MM. T helper 17.1 cells associate with multiple sclerosis disease activity: perspectives for early intervention., 2018, 141(5): 1334–1349.
[19] Paulissen SMJ, van Hamburg JP, Dankers W, Lubberts E. The role and modulation of CCR6+Th17 cell populations in rheumatoid arthritis., 2015, 74(1): 43–53.
[20] Basdeo SA, Cluxton D, Sulaimani J, Moran B, Canavan M, Orr C, Veale DJ, Fearon U, Fletcher JM. Ex-Th17 (nonclassical th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells., 2017, 198(6): 2249–2259.
[21] Tahmasebinia F, Pourgholaminejad A. The role of Th17 cells in auto-inflammatory neurological disorders., 2017, 79(Pt B): 408–416.
[22] Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system., 2019, 4(37): eaav0492.
[23] Profaci CP, Munji RN, Pulido RS, Daneman R. The blood- brain barrier in health and disease: important unanswered questions., 2020, 217(4): e20190062.
[24] Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity., 2013, 13(9): 666–678.
[25] Breuer J, Korpos E, Hannocks MJ, Schneider-Hohendorf T, Song J, Zondler L, Herich S, Flanagan K, Korn T, Zarbock A, Kuhlmann T, Sorokin L, Wiendl H, Schwab N. Blockade of MCAM/CD146 impedes CNS infiltration of T cells over the choroid plexus., 2018, 15(1): 236.
[26] Cipollini V, Anrather J, Orzi F, Iadecola C. Th17 and cognitive impairment: possible mechanisms of action., 2019, 13: 95.
[27] Haqqani AS, Stanimirovic DB. Intercellular interactomics of human brain endothelial cells and th17 lymphocytes: a novel strategy for identifying therapeutic targets of CNS inflammation., 2011, 2011: 175364.
[28] Witowski J, Pawlaczyk K, Breborowicz A, Scheuren A, Kuzlan-Pawlaczyk M, Wisniewska J, Polubinska A, Friess H, Gahl GM, Frei U, Jörres A. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells., 2000, 165(10): 5814–5821.
[29] Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CRW. Cellular mechanisms of IL-17-induced blood-brain barrier disruption., 2010, 24(4): 1023–1034.
[30] Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, Suzumura A. Production and functions of IL-17 in microglia., 2008, 194(1–2): 54–61.
[31] Debnath M, Berk M. Th17 pathway-mediated immunopathogenesis of schizophrenia: mechanisms and implications., 2014, 40(6): 1412–1421.
[32] Michinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage., 2019, 20(3): 571.
[33] Meares GP, Ma XY, Qin HW, Benveniste EN. Regulation of CCL20 expression in astrocytes by IL-6 and IL-17., 2012, 60(5): 771–781.
[34] Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, Fitzgerald DC, Shindler KS, Rostami A. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis., 2009, 6: 14.
[35] Kang ZZ, Altuntas CZ, Gulen MF, Liu CN, Giltiay N, Qin HW, Liu LP, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li XX. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis., 2010, 32(3): 414–425.
[36] Williams JL, Manivasagam S, Smith BC, Sim J, Vollmer LL, Daniels BP, Russell JH, Klein RS. Astrocyte-T cell crosstalk regulates region-specific neuroinflammation., 2020, 68(7): 1361–1374.
[37] Tzartos JS, Craner MJ, Friese MA, Jakobsen KB, Newcombe J, Esiri MM, Fugger L. IL-21 and IL-21 receptor expression in lymphocytes and neurons in multiple sclerosis brain., 2011, 178(2): 794–802.
[38] Birkner K, Wasser B, Ruck T, Thalman C, Luchtman D, Pape K, Schmaul S, Bitar L, Krämer-Albers EM, Stroh A, Meuth SG, Zipp F, Bittner S. β1-Integrin- and KV1.3 channel-dependent signaling stimulates glutamate release from Th17 cells., 2020, 130(2): 715–732.
[39] Williams PR, Marincu BN, Sorbara CD, Mahler CF, Schumacher AM, Griesbeck O, Kerschensteiner M, Misgeld T. A recoverable state of axon injury persists for hours after spinal cord contusion in vivo., 2014, 5: 5683.
[40] Wu B, Wan YS. Molecular control of pathogenic Th17 cells in autoimmune diseases., 2020, 80: 106187.
[41] Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, Garefalaki A, Potocnik AJ, Stockinger B. Fate mapping of IL-17-producing T cells in inflammatory responses., 2011, 12(3): 255–263.
[42] Jain R, Chen Y, Kanno Y, Joyce-Shaikh B, Vahedi G, Hirahara K, Blumenschein WM, Sukumar S, Haines CJ, Sadekova S, McClanahan TK, McGeachy MJ, O'Shea JJ, Cua DJ. Interleukin-23-induced transcription factor Blimp-1 promotes pathogenicity of T helper 17 cells., 2016, 44(1): 131–142.
[43] Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1., 2013, 496(7446): 513–517.
[44] Haase S, Wilck N, Kleinewietfeld M, Müller DN, Linker RA. Sodium chloride triggers Th17 mediated autoimmunity., 2019, 329: 9–13.
[45] Fleetwood AJ, Cook AD, Hamilton JA. Functions of granulocyte-macrophage colony-stimulating factor., 2005, 25(5): 405–428.
[46] El-Behi M, Ciric B, Dai H, Yan YP, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF., 2011, 12(6): 568–575.
[47] Fujio K, Komai T, Inoue M, Morita K, Okamura T, Yamamoto K. Revisiting the regulatory roles of the TGF-β family of cytokines., 20 16, 15(9): 917– 922.
[48] Bettelli E, Carrier Y, Gao WD, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells., 2006, 441(7090): 235–238.
[49] Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation., 2011, 34(3): 396–408.
[50] Mangan PR, Harrington LE, O'Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage., 2006, 441(7090): 231–234.
[51] Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation., 2007, 26(5): 579–591.
[52] Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen WJ, O'Shea JJ. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling., 2010, 467(7318): 967–971.
[53] Chikuma S, Suita N, Okazaki IM, Shibayama S, Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo., 2012, 13(6): 596–603.
[54] Wang HX, Zhu DH. Functions of smad and its binding proteins in TGF β signal pathway., 2003, 25(4): 479–483.
王海霞, 朱大海. Smad及其结合蛋白在TGF β信号传导中的功能. 遗传, 2003, 25(4): 479–483.
[55] Morianos I, Papadopoulou G, Semitekolou M, Xanthou G. Activin-A in the regulation of immunity in health and disease., 2019, 104: 102314.
[56] Zhang S, Takaku M, Zou LY, Gu AD, Chou WC, Zhang G, Wu B, Kong Q, Thomas SY, Serody JS, Chen X, Xu XJ, Wade PA, Cook DN, Ting JPY, Wan YY. Reversing SKI-SMAD4-mediated suppression is essential for TH17 cell differentiation., 2017, 551(7678): 105–109.
[57] Wu B, Zhang S, Guo ZL, Bi YM, Zhou MX, Li P, Seyedsadr M, Xu XJ, Li JL, Markovic-Plese S, Wan YY. The TGF-β superfamily cytokine activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation., 2021, 54(2): 308–323.e6.
[58] Liu HP, Yao SX, Dann SM, Qin HW, Elson CO, Cong YZ. ERK differentially regulates Th17- and Treg-cell development and contributes to the pathogenesis of colitis., 2013, 43(7): 1716–1726.
[59] Brereton CF, Sutton CE, Lalor SJ, Lavelle EC, Mills KHG. Inhibition of ERK MAPK suppresses IL-23- and IL-1- driven IL-17 production and attenuates autoimmune disease., 2009, 183(3): 1715–1723.
[60] Xu Q, Jin XX, Zheng MZ, Rohila D, Fu GT, Wen ZY, Lou J, Wu SQ, Sloan R, Wang L, Hu H, Gao X, Lu LR. Phosphatase PP2A is essential for TH17 differentiation., 2019, 116(3): 982–987.
[61] Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function., 2019, 47(D1): D155–D162.
[62] Baumjohann D, Ansel KM. MicroRNA-mediated regulation of T helper cell differentiation and plasticity., 2013, 13(9): 666–678.
[63] Chen C, Zhou YF, Wang JQ, Yan YP, Peng LS, Qiu W. Dysregulated microRNA involvement in multiple sclerosis by induction of T helper 17 cell differentiation., 2018, 9: 1256.
[64] Ichiyama K, Gonzalez-Martin A, Kim BS, Jin HY, Jin W, Xu W, Sabouri-Ghomi M, Xu SB, Zheng P, Xiao CC, Dong C. The microRNA-183-96-182 cluster promotes T helper 17 cell pathogenicity by negatively regulating transcription factor Foxo1 expression., 2016, 44(6): 1284–1298.
[65] Bamodu OA, Huang WC, Lee WH, Wu A, Wang LS, Hsiao M, Yeh CT, Chao TY. Aberrant KDM5B expression promotes aggressive breast cancer through MALAT1 overexpression and downregulation of hsa-miR-448., 2016, 16: 160.
[66] Hong XH, Xu Y, Qiu XF, Zhu YK, Feng X, Ding ZJ, Zhang SF, Zhong LF, Zhuang YF, Su C, Hong XY, Cai JC. MiR-448 promotes glycolytic metabolism of gastric cancer by downregulating KDM2B., 2016, 7(16): 22092–22102.
[67] Lou Q, Liu RX, Yang XL, Li WQ, Huang LL, Wei LL, Tan HL, Xiang NL, Chan K, Chen JX, Liu HL. MiR-448 targets IDO1 and regulates CD8+T cell response in human colon cancer., 2019, 7(1): 210.
[68] Sasahira T, Kurihara M, Nishiguchi Y, Fujiwara R, Kirita T, Kuniyasu H. NEDD 4 binding protein 2-like 1 promotes cancer cell invasion in oral squamous cell carcinoma., 2016, 469(2): 163–172.
[69] Wu RH, He QY, Chen HT, Xu M, Zhao N, Xiao Y, Tu QQ, Zhang WJ, Bi XY. MicroRNA-448 promotes multiple sclerosis development through induction of Th17 response through targeting protein tyrosine phosphatase non-receptor type 2 (PTPN2)., 2017, 486(3): 759–766.
[70] Keller A, Leidinger P, Lange J, Borries A, Schroers H, Scheffler M, Lenhof HP, Ruprecht K, Meese E. Multiple sclerosis: microRNA expression profiles accurately differentiate patients with relapsing-remitting disease from healthy controls., 2009, 4(10): e7440.
[71] Zhu ED, Wang X, Zheng B, Wang Q, Hao JL, Chen SM, Zhao Q, Zhao LQ, Wu ZZ, Yin ZN. MiR-20b suppresses Th17 differentiation and the pathogenesis of experimental autoimmune encephalomyelitis by targeting RORγt and STAT3., 2014, 192(12): 5599–5609.
[72] Chen J, Martindale JL, Abdelmohsen K, Kumar G, Fortina PM, Gorospe M, Rostami A, Yu SG. RNA-binding protein HuR promotes Th17 cell differentiation and can be targeted to reduce autoimmune neuroinflammation., 2020, 204(8): 2076–2087.
[73] Chen J, Martindale JL, Cramer C, Gorospe M, Atasoy U, Drew PD, Yu SG. The RNA-binding protein HuR contributes to neuroinflammation by promoting C-C chemokine receptor 6 (CCR6) expression on Th17 cells., 2017, 292(35): 14532–14543.
[74] Roy DG, Chen J, Mamane V, Ma EH, Muhire BM, Sheldon RD, Shorstova T, Koning R, Johnson RM, Esaulova E, Williams KS, Hayes S, Steadman M, Samborska B, Swain A, Daigneault A, Chubukov V, Roddy TP, Foulkes W, Pospisilik JA, Bourgeois-Daigneault MC, Artyomov MN, Witcher M, Krawczyk CM, Larochelle C, Jones RG. Methionine metabolism shapes T helper cell responses through regulation of epigenetic reprogramming., 2020, 31(2): 250–266.e9.
[75] Cho JJ, Xu ZW, Parthasarathy U, Drashansky TT, Helm EY, Zuniga AN, Lorentsen KJ, Mansouri S, Cho JY, Edelmann MJ, Duong DM, Gehring T, Seeholzer T, Krappmann D, Uddin MN, Califano D, Wang RL, Jin L, Li HM, Lv DW, Zhou DH, Zhou L, Avram D. Hectd3 promotes pathogenic Th17 lineage through Stat3 activation and Malt1 signaling in neuroinflammation., 2019, 10(1): 701.
[76] Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen- colour flow cytometry: unravelling the immune system., 2004, 4(8): 648–655.
[77] Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Raychowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, Trombetta JJ, Gennert D, Gnirke A, Goren A, Hacohen N, Levin JZ, Park H, Regev A. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells., 2013, 498(7453): 236–240.
[78] Gaublomme JT, Yosef N, Lee Y, Gertner RS, Yang LV, Wu C, Pandolfi PP, Mak T, Satija R, Shalek AK, Kuchroo VK, Park H, Regev A. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity., 2015, 163(6): 1400–1412.
Research progress on the role and regulatory mechanism of pathogenic Th17 cells in neuroinflammation
Hongyu Dai1,2, Dong Ji1,2, Cheng Tan1,2, Jie Sun3, Hao Yao1,2
Neuroinflammation is a complex immune response in the central nervous system against various factors such as injury, infection and toxins which interfere with homeostasis, involving a variety of immune cells lingering in the central nervous system. Persistent neuroinflammation is a common denominator of the etiology and course of all neurological diseases, including neurodevelopmental, neurodegenerative and psychiatric disorders, such as Alzheimer’s disease, Parkinson’s disease, multiple sclerosis and depression. Th17 cells, known as an important subtpye of CD4+T cells, mediate immune responses against extracellular bacteria and fungi in steady-state and maintain the defense function of the intestinal mucosal barrier. However, when the cytokine microenvironmentundergoes inflammatory changes, Th17 cells can transform into a highly pro-inflammatory pathogenic phenotype, break through the blood-brain barrier and recruit more inflammatory cells to participate in neuroinflammation, ultimately leading to neurodegeneration. In this review, we summarize the differentiation regulation of pathogenic Th17 cells and their roles in neuroinflammation, which is informative for understanding the interactions between immune system and nervous system.
neuroinflammation; pathogenic Th17 cells; blood-brain barrier; RORγt
2022-02-12;
2022-03-18;
2022-03-25
江苏省科技厅省级重点研发计划(社会发展)项目(编号:SBE2021741263)和南京医科大学第二附属医院789人才培养计划(编号:789ZYRC080236)资助[Supported by the Provincial Key R&D Program (Social Development) of Science and Technology Department of Jiangsu Province (No. SBE2021741263), and the 789 Talents Training Program of the Second Affiliated Hospital of Nanjing Medical University (No. 789ZYRC080236)]
戴鸿宇,在读硕士研究生,专业方向:麻醉学。E-mail: daihongyu@njmu.edu.cn
姚昊,博士,副教授,研究方向:围术期脏器保护。E-mail: yaohao@njmu.edu.cn
孙杰,博士,副教授,研究方向:麻醉与术后认知功能。E-mail: dgsunjie@hotmail.com
10.16288/j.yczz.22-030
(责任编委: 何淑君)
猜你喜欢
现代临床医学(2021年4期)2021-07-31
神经损伤与功能重建(2020年10期)2020-12-23
神经损伤与功能重建(2020年11期)2020-12-01
中成药(2017年12期)2018-01-19
浙江农业科学(2016年11期)2016-05-04
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国畜牧兽医文摘(2015年9期)2015-12-29
飞碟探索(2015年11期)2015-09-10
中国生化药物杂志(2015年4期)2015-07-07
医学研究杂志(2015年12期)2015-06-10
