
高通量测序分析PIK3CA突变型与野生型结直肠癌的肠道菌群特征*
2022-04-15 08:45尚付梅任中海万里新张敬伟李长生马慧利张蕾冀叶袁小笋刘红利
中国肿瘤临床 2022年6期
尚付梅 任中海 万里新 张敬伟 李长生 马慧利 张蕾 冀叶 袁小笋 刘红利
结直肠癌是全球第三大常见的恶性肿瘤,每年约860 000例患者死于该病,在中国,结直肠癌已成为恶性肿瘤相关死亡率的第五大诱因,其发病率预计将会继续增长[1]。PIK3CA是结直肠癌中最常见的突变癌基因之一。据报道,约有15%~20%的结直肠癌患者携带PIK3CA突变[2]。近年来,越来越多的研究报道肠道菌群与慢性肝病、肠易激综合征、炎症性肠病、结直肠癌等一系列疾病密切相关[3-4]。肠道菌群的组成结构及多样性变化对多系统疾病的发生和发展起重要作用。目前,尚缺乏报道PIK3CA突变型与野生型结直肠癌患者之间的肠道菌群结构。因此,本研究采用16s DNA 高通量测序技术,分析PIK3CA不同基因型结直肠癌患者之间的肠道菌群结构,揭示PIK3CA基因状态与结直肠癌患者肠道菌群的关系,为探究肠道菌群对PIK3CA基因状态的影响提供潜在的科学依据。
1 材料与方法
1.1 临床资料
选取2016年12月至2017年12月华中科技大学同济医学院附属协和医院饮食结构相似的局部可切除且结直肠癌患者19例,收集所有患者手术切除的肿瘤黏膜组织,采用实时荧光定量PCR(ARMS)技术检测PIK3CA的表达状态,根据PIK3CA检测结果分为两组:PIK3CA突变型组(n=4),PIK3CA野生型组(n=15)。纳入标准:1)饮食结构相似,无特殊饮食习惯;2)局部可手术切除的原发结直肠癌患者。排除标准:1)其它部位恶性肿瘤病史患者;2)继发结直肠癌患者;3)多部位原发结直肠癌患者;4)术前6天行肠道准备;5)术前3个月使用过抗生素;6)术前行新辅助放疗或化疗;7)存在其它胃肠道系统性疾病;8)术前2周内使用过益生菌或益生元;9)存在代谢性或感染性疾病。入组患者的PIK3CA突变型率为21.05%;平均年龄为(62.68±13.31)岁。
1.2 方法
1.2.1 样本采集 所有患者签署知情同意书后接受结直肠癌肿瘤切除术,并留取肿瘤组织活检标本,迅速置入无菌试剂盒中,并用冰盒将其转运至实验室,再迅速转移至-80℃冰箱冻存。所有操作均在无菌条件下进行,避免环境细菌污染及交叉污染。
1.2.2 基因组 DNA 提取及PCR扩增 采用Mag-Bind Soil DNA Kit试剂盒(购自美国Omega 公司),提取所有患者的基因组DNA,并使用0.8% 琼脂糖凝胶电泳检测DNA纯度及浓度。使用PCR扩增仪(购自美国Applied Biosystems公司)对标准细菌的16S rDNA V3V4可变区进行PCR扩增,其上、下游引物序列分别为:5’-ACTCCTACGGGAGGCAGCA-3’,5’-GGACTACHVGGGTWTCTAAT-3’。
1.2.3 测序文库构建及上机高通量测序 采用TruSeq Nano DNA LT Library Prep Kit 试剂盒构建高通量测序文库,并在 Illumina MiSeq PE 250(购自美国Illumina公司)平台上执行2×300 bp的双端、高通量测序。
1.2.4 生物信息学分析 使用QIIME软件对原始数据进行整理、过滤及质量评估,得到有效的测序数据。使用usearch软件,通过UPARSE-OTU algorithm,对获得的有效数据进行聚类,以97%的序列相似性进行分类单元(OTUs)聚类,然后对OTUs 的代表序列进行物种注释,得到对应的物种信息和基于物种的丰度分布情况。基因OTU丰度矩阵,采用R软件计算各组所共有和特有的OTU数量,绘制Venn图。根据OTU划分和分类地位鉴定结果,用 R软件分析比较不同分组在不同分类水平上的组成和丰度分布。通过主成分分析(principal component analysis,PCA)对群落数据结构进行自然分解并通过对样本排序,从而观测样本之间的差异。 Metastats分析是基于群落丰度采用Mothur 软件分析样本(组)间分类学组成差异的统计学算法,对各个分类单元在样本(组)之间的丰度差异进行两两比较检验。使用Mothur软件,计算丰度位于前50位的优势属之间的Spearman等级相关系数,构建优势微生物类群的关联网络。
1.3 统计学分析
采用SPSS 22.0软件进行统计学分析,采用Chi-Square检验、t检验、Fisher 精确检验进行组间数据比较分析,采用R软件统计作图。对于α多样性指数(Chao1、ACE、Shannon和Simpson)使用Wilcoxon秩和检验比较各组样本微生物多样性差异。使用变异的多元方差分析(permutational multivariate analysis of variance,PERMANOVA)来评估两组之间的微生物群落结构差异。以P<0.05为差异具有统计学意义。
2 结果
2.1 16S rDNA高通量测序结果
本研究通过对PIK3CA野生型与突变型结直肠癌组织中16S rDNA 基因的V3-V4区进行高通量测序,共获得高质量的优化序列共251 211 911条,每个样品中的平均序列数为39 004条,平均序列长度为429 bp,且这些序列长度主要集中在400 bp 以上,本研究的测序深度基本趋于饱和状态。
本研究通过UCLUST序列比对方法对OTU进行聚类分析,以97%的序列相似度作为OTU划分阈值,并将每个OTU中丰度最高的序列定义为该OTU的代表序列,分析每个样本在各分类水平上(phylum,class,order,family,genus,species)的微生物群落鉴定结果。根据OTU丰度矩阵,计算各组所共有和特有的OTU数量,即 Venn图(图1A),有190个OTU在PIK3CA野生型与突变型肿瘤组织中是共有的,有88个OTU在PIK3CA突变型结直肠癌肿瘤组织中特有(图1A)。
2.2 PIK3CA突变型与野生型分组之间的菌群组成结构
根据OTU分类结果,对PIK3CA突变型与野生型之间的菌群组成结构进行分析,在门(phylum)水平上,PIK3CA突变型与野生型肿瘤组织中的优势菌门主要均由变形菌门(Proteobacteria)、栖热菌门(Thermi)、放线菌门(Actinobacteria)、厚壁菌门(Firmicutes)和拟杆菌门(Bacteroidetes)组成,占总群落的98%以上,其中占比最高的是变形菌门,占比70%以上(图1B)。

图1 PIK3CA突变型与野生型分组
2.3 PIK3CA突变型与野生型分组之间的Alpha多样性分析
Alpha多样性反映微生物群落的丰富度和均匀度,常用的度量Alpha多样性的度量标准包括:ACE指数,Chao1指数,Simpson指数和Shannon指数。Chao1或ACE指数越大,表明群落的丰富度越高;Shannon指数则综合考虑了群落的丰富度和均匀度,Shannon指数值越高,表明群落的多样性越高。Shannon指数对群落的丰富度以及稀有OTU更敏感,Simpson指数值越高,表明群落多样性越高。本研究采用Wilcoxon秩和检验比较了PIK3CA突变型与野生型之间微生物菌群多样性差异,研究结果显示ACE指数、Chao1指数、Shannon指数和Simpson指数在PIK3CA突变型的结直肠癌患者肿瘤组织中明显高于PIK3CA野生型(图1C,均P<0.001),说明PIK3CA突变型与PIK3CA野生型的结直肠癌患者肿瘤组织中的菌群多样性有显著性差异,揭示了PIK3CA基因表达状态可能与结直肠癌肿瘤局部微环境的菌群多样性密切相关。
2.4 PIK3CA突变型与野生型之间的Beta多样性分析
Beta多样性分析主要考察不同样本之间群落结构的差异性。通过PCA分析反映不同分组之间的群落组成差异。本研究采用PCA分析比较了结直肠癌肿瘤组织中PIK3CA突变型与野生型之间的菌群结构差异,使用PERMANOVA分析两组之间的统计学关系,结果显示PIK3CA突变型与野生型的结直肠癌肿瘤组织中的菌群结构有显著性差异(图2,P=0.004)。

图2 PIK3CA突变型与野生型分组之间菌群结构的PCA分析
2.5 PIK3CA突变型与野生型之间的Metastats分析
本研究采用Metastats分析比较了结直肠癌肿瘤组织中PIK3CA突变型与野生型之间的微生物群落组成差异。Metastats结果显示,在门水平上,有5种菌门存在显著差异,其中厚壁菌门(Firmicutes),装甲菌门(Armatimonadetes),放线菌门(Actinobacteria)及拟杆菌门(Bacteroidetes)4种菌门在PIK3CA突变型肠癌患者肿瘤组织中明显富集,而变形菌门(Proteobacteria)在PIK3CA野生型患者中明显富集(图3,P<0.01)。在属(genus)水平上,有29种菌属有显著性差异(本研究中仅列出差异最显著的前20个分类单元),其中包含Allobaculum,拟无枝酸菌(Amycolatopsis)及厌氧芽孢杆菌属(Anoxybacillus)在内的18种菌属在PIK3CA突变型患者肿瘤组织明显富集,而不动杆菌属(Acinetobacter)和鞘氨醇单胞菌属(Sphingomonas)在PIK3CA野生型患者中明显富集(图4,P<0.01)。

图3 门水平上,PIK3CA突变型与野生型结直肠癌肿瘤组织之间的菌门丰度分布

图4 属水平上,PIK3CA突变型与野生型结直肠癌肿瘤组织之间的菌属丰度分布
2.6 PIK3CA突变型与野生型分组之间的Spearman关联网络分析
Spearman关联网络分析根本目的是考察不同群落成员之间的相互作用,通过关联分析的方法,找寻群落成员在不同生境下共同出现或彼此排斥的相互作用模式,从而推断不同微生物类群之间可能的相互“协作”或“竞争”关系。根据OTU或各分类单元在不同样本中的丰度分布,可以寻找彼此之间呈现正相关或负相关的微生物类群,进而构建优势微生物类群的关联网络,探索其彼此相关的生态学意义。如图5所示,红线表明正相关,绿线表明负相关。通过某节点的连接越多,表明该属与菌群中其它成员的关联越多。在PIK3CA野生型结直肠癌肿瘤组织中,本研究发现双歧杆菌属(Bifidobacterium)与假单胞菌(Pseudomonas)呈正相关(图5A)。在PIK3CA突变型的结直肠癌肿瘤组织中,拟杆菌属(Bacteroides)与嗜热脂肪地芽孢杆菌(Geobacillus)、蛭弧菌(Bdellovibrio)、棒杆菌属(Corynebacterium)呈正相关(图5B)。
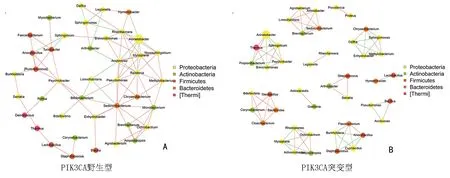
图5 PIK3CA突变型与野生型结直肠癌肿瘤组织之间的关联网络分析
3 讨论
本研究发现结直肠癌患者PIK3CA突变型与野生型中的肿瘤组织微生物菌群,无论是Alpha多样性,还是Beta多样性均有显著性差异,首次揭示了不同PIK3CA基因状态下结直肠癌患者肠道菌群组成结构不同。
近期有研究报道了肠道菌群与结直肠癌患者的发生及不良预后相关[5]。产肠毒素脆弱类杆菌(Bacteroides fragilis)是Bacteroidetes成员中的一种常见的人类共生菌。在动物实验中,有学者发现Bacteroides fragilis通过激活STAT3的表达,分泌促炎性细胞因子 IL-17,进而诱导结肠癌的发生[6]。在人体实验中,有研究发现,Bacteroides fragilis在结直肠癌患者黏膜组织中明显富集[7],并且与其较晚的临床分期显著相关,说明肠黏膜中Bacteroides fragilis的丰度越高,其临床预后越差[8]。此外,Viljoen 等[7]也发现Bacteroides fragilis与结直肠癌的不良预后有关。另有研究发现,在人类结直肠癌的原发肿瘤部位中可检出Bacteroides,同样在肿瘤远处转移部位中亦可检出同样的微生物菌,说明Bacteroides在肿瘤原发灶和转移瘤中保持了微生物组的稳定性,揭示了Bacteroides在肿瘤转移过程中的重要地位[9]。本研究发现Bacteroidetes在PIK3CA突变型肠癌患者肿瘤组织明显富集,在PIK3CA野生型患者中并未观察到Bacteroidetes的增多,推测肠道菌群中高丰度的Bacteroidetes是否促进了PIK3CA突变,进而影响了结直肠癌患者的不良预后,这有待于在未来的研究中进一步揭示。
众所周知,PIK3CA编码Ⅰ类磷脂酰肌醇-3-激酶(phosphatidylino-sitol 3-kinases,PI3Ks)的p110催化亚单位。目前对PI3Ks的研究主要集中在Ⅰ型。Ⅰ型由催化亚基和调节亚基构成的,活化的RTKs与调节亚基结合后使催化亚基到达细胞膜,活化Ⅰ型PI3Ks,激活PI3K-AKT-mTOR信号转导通路,影响细胞的增殖、分化,进而导致人类各种恶性肿瘤的发生[10],比如在乳腺癌,膀胱癌和结直肠癌等多种恶性肿瘤中可观察到PIK3CA的高突变频率[11]。PIK3CA突变不仅可以减少细胞凋亡,同时还可以促进肿瘤浸润、提高其下游激酶PI3Ks的活性[12]。PIK3CA是结直肠癌中最常见的突变癌基因之一。据报道,约有15%~20%的结直肠癌患者携带PIK3CA突变[2]。本研究发现PIK3CA在结直肠癌中的突变率为21.05%,不同研究中报道的PIK3CA突变率有所差异,这可能与种族人群、研究队列和检测技术有关[13]。一些研究表明,PIK3CA突变与结直肠癌患者的不良预后密切相关[14]。Farina等[15]发现具有PIK3CA外显子20突变的患者比PIK3CA野生型肠癌患者具有更低的无病生存期。Ogino等[16]报道了PIK3CA突变是Ⅰ~Ⅲ期结直肠癌患者的不良预后因素。本研究发现,Bacteroides fragilis与PIK3CA突变均与结直肠癌患者的不良预后密切相关,同时也发现PIK3CA突变型的结直肠癌患者中Bacteroidetes明显富集。既往研究发现PIK3CA突变会引起PI3K/AKT/mTOR通路的激活,而mTOR是重要的代谢调控通路,影响多种代谢产物的合成[10]。Ogino等[16]发现结直肠癌患者中的氨基酸、无机酸、醛、脂肪酸等多种代谢产物与对肠道菌群结构产生直接的影响。因此,本研究推测,PIK3CA突变是否潜在的引起了肠道菌群结构的变化,进一步缩短了结直肠癌患者的生存期。该研究同时发现益生菌干预显著改善大肠癌患者肠道菌群结构,并特异性降低包含Fusobacterium在内的某些致病菌丰度,初步显示出益生菌干预对大肠癌患者肠道菌群结构的调整能力。此外,该研究也发现益生菌通过调节免疫功能和抑制潜在有害细菌的生长等机制预防结直肠癌的发生[16]。因此,本研究认为,未来的研究中,能否通过肠道菌群移植,益生菌干预,寻找靶向于特定肠道菌群的药物等一系列途径,来逆转肠道菌群稳态的破坏,进而阻断肠道菌群变化给人类带来的包括结直肠癌等在内的一系列疾病,期待更多的研究进一步揭示。
综上所述,本研究发现PIK3CA突变型与野生型结直肠癌患者肿瘤组织中的微生物群落结构有显著性差异,揭示了结直肠癌患者中的肠道菌群结构与PIK3CA突变之间存在着潜在的关系,然而其中具体的机制尚不明确,有待更深入的研究进一步揭示。
猜你喜欢
当代水产(2022年8期)2022-09-20
今日农业(2022年14期)2022-09-15
中老年保健(2022年2期)2022-08-24
中国饲料(2022年5期)2022-04-26
昆明医科大学学报(2022年3期)2022-04-19
昆明医科大学学报(2022年2期)2022-03-29
现代仪器与医疗(2021年4期)2021-11-05
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年5期)2021-07-22
学校教育研究(2020年7期)2020-04-09
