
加味茵陈蒿口服液的质量控制研究
2022-04-11 03:33:34郭振环吕一舟张志强李向辉刘永录范云鹏
动物医学进展 2022年4期
郭振环,吕一舟,马 霞,张志强,李向辉,刘永录,范云鹏
(1.河南牧业经济学院,河南郑州 450046;2.河南省康星药业股份有限公司,河南郑州 451464;3.西北农林科技大学,陕西杨凌 712100)
《中国兽药典》2015年版二部收载的茵陈蒿散[1],源于张仲景的《伤寒论》,由茵陈、栀子、大黄组成。有清热解毒、利胆、退黄等功效[2-3],临床常用于病毒性肝炎、黄疸性肝炎、胆汁淤积等湿热内蕴证[4-6]。加味茵陈蒿口服液是在茵陈蒿散基础上加味甘草,新方剂以茵陈为君药,可清除肝胆湿热;栀子为臣药,可清泄三焦湿热;大黄为佐药,可荡涤肠胃瘀热;甘草为使药,益气补脾,止痛,有调和诸药之功效。前3味药均有苦寒之性,可清除湿热,而甘草性平,可调和苦寒之性,解栀子苷毒性[7],还可调节口味,制成口服液便于临床使用。
目前《中国兽药典》收载的茵陈蒿散中只有显微鉴别和栀子、大黄的薄层色谱鉴别[1],没有相应的含量测定方法。本试验对加味茵陈蒿口服液进行初步质量控制标准研究,从口服液的相对密度、pH、薄层色谱法(thin layer chromatography,TLC)定性鉴别、再到高效液相色谱法(high performance liquid chromatography,HPLC)定量检测,以期为口服液的进一步开发提供基础依据。
1 材料与方法
1.1 材料
1.1.1 主要试剂 茵陈、栀子、大黄、甘草均为亳州药强中药材有限公司产品;栀子苷(MUST-19102310)、甘草酸单铵盐对照品(MUST-19073110),均为成都曼斯特生物科技有限公司产品;大黄素对照品(CDAA-281250),上海安谱实验科技股有限公司产品;乙醚、乙酸乙酯、正己烷、正丁醇、丙酮、甲酸、冰醋酸等为分析纯,均为山东双双化工有限公司产品;甲醇、乙腈、磷酸为色谱纯,均为天津市康科德科技有限公司产品。
1.1.2 主要仪器 高效液相色谱仪(agilent technologies 1260),安捷伦科技中国有限公司产品;超声波清洗器(SYU-3-100D),郑州生元仪器有限公司产品;万分之一天平(BSA224S),赛多利斯科学仪器有限公司产品;暗箱四用紫外分析仪(ZF-8),上海勤科分析仪器有限公司产品;雷磁酸度计(PHS-3C),上海仪电科学仪器股份有限公司产品;循环水真空泵(SHZ-D)、低温冷却液循环泵(DLSB-5/10)、旋转蒸发器(RE-5299),均为郑州科达机械仪器设备有限公司产品。
1.2 方法
1.2.1 加味茵陈蒿口服液及阴性药液的制备
(1)加味茵陈蒿口服液制备:取茵陈120 g,栀子60 g,大黄45 g,甘草25 g,按照前期试验中确定的最佳制备工艺进行10批口服液的制备:补足2倍吸水量后加12倍水,浸泡0.5 h,煎煮2次,每次1 h,2次药液合并后浓缩至1 mL相当于原药材1g。
(2)阴性药液制备:按上述工艺分别制备缺栀子、缺大黄、缺甘草的阴性对照药液。
1.2.2 口服液的一般检查
(1)相对密度的测定:取洁净、干燥并精密称定重量的比重瓶,装满测试样品;装上温度计(瓶中应无气泡),置20℃的水浴中放置10 min~20 min,使瓶内被测物的温度达到20℃;用滤纸除去溢出侧管的液体,立即盖上罩;将比重瓶自水浴中取出,用滤纸将比重瓶的外面擦干,精密称定,减去比重瓶的重量,求得供试品的重量;将供试品倾去,洗净比重瓶,并装满新煮沸过的冷水,照上法测得同一温度时水的重量,按下式计算,即得。
样品的相对密度=供测试样品重量/水重量
(2)pH的测定:采用酸度计检测每批样品pH。
1.2.3 薄层色谱法鉴别
(1)栀子的鉴别:精密量取浓缩液3 mL,加乙醚20 mL,超声20 min,弃乙醚后挥干溶剂,再加入乙酸乙酯30 mL,加热回流1 h,放冷后取乙酸乙酯层蒸干,残渣加甲醇1 mL溶解,滤过,滤液作为样品溶液。同法制得栀子对照药材和缺栀子的阴性对照溶液。取栀子苷对照品适量,加甲醇制成每 1 mL 含0.3 mg的栀子苷对照品溶液。吸取上述4种溶液各2 μL分别点于同一硅胶G板上,以乙酸乙酯-丙酮-水(5∶5∶0.6)为展开剂展开,取出晾干后喷100 g/L的硫酸乙醇溶液,加热至105℃使薄层板上有斑点显色。
(2)大黄的鉴别:精密量取浓缩液3 mL,加甲醇25 mL,加热回流1 h,放冷滤过后把滤液蒸干。残渣加水5 mL溶解,再加盐酸1 mL,水浴加热30 min立即冷却。用乙醚提取2次,每次5 mL,合并乙醚液,蒸干。残渣加三氯甲烷1 mL使溶解,作为供试品溶液。同法制得大黄对照药材和缺大黄的阴性对照溶液。取大黄素对照品适量,加甲醇制成每 1 mL 含0.8 mg的大黄素对照品溶液。吸取上述4种溶液各2 μL分别点于同一硅胶GF254板上,正己烷-乙酸乙酯-甲酸(30∶10∶0.5)为展开剂展开,取出晾干后置于365 nm紫外灯下观察。
(3)甘草酸铵的鉴别:精密量取浓缩液2 mL,加乙醚8 mL,加热回流1 h,弃乙醚后蒸干溶液,药渣加甲醇6 mL,加热回流1 h,滤过后滤液蒸干;残渣加水8 mL溶解,正丁醇提取3次,每次4 mL,合并正丁醇液;用水洗涤3次,弃去水液,正丁醇液蒸干,残渣加甲醇1 mL使溶解作为供试品溶液。同法制得甘草对照药材和缺甘草的阴性对照溶液。取甘草酸铵对照品适量,加甲醇制成每 1 mL 含2.0 mg的甘草酸铵对照品溶液。吸取上述4种溶液各2 μL分别点于同一硅胶GF254板上,正丁醇-冰醋酸-水(6∶1∶3)为展开剂展开,取出晾干后置于254 nm紫外灯下观察。
1.2.4 含量测定
(1)色谱条件:栀子苷的流动相为乙腈∶1 mL/L磷酸水(15∶85),检测波长为238 nm;大黄素流动相为甲醇-1 mL/L磷酸水(85∶15),检测波长为254 nm;甘草酸单铵盐流动相为乙腈-1 mL/L磷酸水(37∶63),检测波长为250 nm。色谱柱均为C18柱(4.6×250 mm,5 μm),流速1 mL/min,柱温30℃,进样10 μL。
(2)供试品溶液和阴性对照溶液的制备:
①精密量取浓缩液0.5 mL,用甲醇溶液定容至 10 mL,超声处理30 min。放冷,摇匀滤过,取滤液,作为测定栀子苷的供试品溶液。
②精密量取浓缩液1 mL,用甲醇溶液定容至 10 mL,加热回流1 h。放冷,离心后过滤取续滤液蒸干,加80 mL/L盐酸溶液5 mL,超声2 min;在盐酸溶液中加三氯甲烷5 mL,取三氯甲烷层,提取3次合并三氯甲烷液,蒸干,加甲醇1 mL摇匀滤过,取滤液,作为测定大黄素的供试品溶液。
③精密量取浓缩液0.5 mL,用 700 mL/L乙醇定容至 10 mL,超声处理30 min。放冷,摇匀,滤过,取滤液,作为测定甘草酸单铵盐的供试品溶液。
(3)对照品溶液的制备:精密称定对照品适量,加甲醇制成30 μg/mL的栀子苷对照品溶液;加甲醇制成40 μg/mL的大黄素对照品溶液;加700 mL/L乙醇制成0.2 mg/mL的甘草酸单铵盐对照品溶液(甘草酸重量=甘草酸铵重量/1.0207)。
(4)专属性考察:分别吸取供试品溶液、阴性对照溶液及相应的对照品溶液各10 μL,按照上述色谱条件检测。
(5)线性关系考察:精密吸取栀子苷对照品6 、10 、14、16、20、22 μL,按照含量测定方法,依次注入液相色谱仪进行测定。分别精密吸取大黄素和甘草酸铵对照品1、2、4、8、10 μL,按照含量测定方法,依次注入液相色谱仪进行测定。以峰面积为纵坐标,进样量(μg)为横坐标,绘制标准曲线,得回归方程。
(6)精密度试验:精密吸取各对照品溶液10 μL,连续进样6次,测定峰面积,计算RSD值。
(7)稳定性试验:取同一供试品(批号:20200610)溶液10 μL,分别在0、3、6、9、12、24 h进样,分别记录栀子苷、大黄素、甘草酸铵的峰面积,计算RSD值。
(8)加样回收率:取已知栀子苷/大黄素/甘草酸单铵盐含量加味茵陈蒿口服液1 mL(批号:20200610),按照添加对照品的量为供试品有效成分含量的80%、100%、120%分为3组,每组重复3次。按供试品溶液的制备及样品测定项下操作,测定其含量,计算平均回收率和RSD值。
(9)含量限度测定:取实验室小试生产的10批口服液样品进行含量测定,根据试验结果制定各成分含量限度。
2 结果与分析
2.1 口服液的一般检查结果
2.1.1 相对密度 经检测口服液的相对密度为1.06 g/mL~1.10 g/mL。
2.1.2 pH检查 经检测口服液pH为4.87~4.90。
2.2 薄层色谱法鉴别结果
栀子的鉴别结果如图1A显示,供试品色谱中,在与栀子苷对照品、栀子对照药材色谱相对应的位置显示相同颜色斑点,缺栀子阴性对照无干扰。
大黄的鉴别结果如图1B显示,供试品色谱中,在与大黄素对照品、大黄对照药材色谱相对应的位置显示黄色荧光斑点,缺大黄阴性对照无干扰。
甘草的鉴别结果如图1C显示,供试品色谱中,在与甘草酸单铵盐对照品、甘草对照药材色谱相对应的位置显示相同暗褐色斑点,缺甘草阴性对照无干扰。
2.3 含量测定结果
2.3.1 专属性考察结果 专属性考察结果显示加味茵陈蒿口服液色谱峰中栀子苷与杂质分离度较好;有芦荟大黄素、大黄酸、大黄素、大黄酚和大黄素甲醚这五种蒽醌类成分,并且阴性对照中在芦荟大黄素和大黄酸位置上有相同成分出现,这两种成分专属性不好,而大黄素分离度较好,故选取专属性好,且含量多的大黄素作为后续样品测定依据;色谱峰中甘草酸单铵盐与杂质分离完全,分离度大于1.5。阴性样品均无干扰,表明栀子苷、大黄素、甘草酸单铵盐的专属性好。
2.3.2 线性关系考察 栀子苷进样量在0.18~0.66 μg内线性关系良好(r=0.999),其回归方程为γ=2938.8χ+283.45。大黄素进样量在0.04~0.40 μg内线性关系良好(r=0.9998),其回归方程为γ=2420.6χ-7.965。甘草酸单铵盐进样量在0.20~2.00 μg内线性关系良好(r=0.9999),其回归方程为γ=483.11χ-1.1483。
2.3.3 精密度试验 精密吸取各对照品溶液10 μL,连续进样6次,栀子苷对照品峰面积RSD为0.34%;大黄素峰面积RSD为0.29%;甘草酸单铵盐峰面积RSD为0.38%。结果表明仪器精密度良好。
2.3.4 稳定性试验 取供试品(批号:20200610)溶液10 μL,分别在0、3、6、9、12、24 h进样,栀子苷峰面积RSD为0.48%;大黄素峰面积RSD为0.75%;甘草酸单铵盐峰面积RSD为0.95%。结果表明口服液中在24 h内稳定。
2.3.5 加样回收率 栀子苷、大黄素、甘草酸单铵盐平均回收率和RSD值,见表1、2、3。栀子苷平均回收率为95.03%~96.93%,RSD范围在0.25%~0.67%;大黄素平均回收率为95.03%~96.93%,RSD范围在1.39%~2.06%;甘草酸单铵盐平均回收率在95.03%~96.93%,RSD范围在0.33%~0.66%。结果表明方法准确度高。

表1 口服液中栀子苷加样回收率试验结果Table 1 Experimental results of the recovery rate of geniposide in oral liquid
2.3.6 含量限度测定 测得栀子苷含量范围为1.343 mg/mL~1.380 mg/mL,栀子苷含量平均值为1.361 mg/mL,最低限度为1.09 mg/mL;大黄素含量范围为8.625 μg/mL~9.298 μg/mL,大黄素含量平均值为8.903 μg/mL,最低限度为7.12 μg/mL;甘草酸单铵盐含量范围为0.875 mg/mL~0.960 mg/mL,甘草酸单铵盐含量平均值为0.929 mg/mL,最低限度为0.73 mg/mL。
3 讨论
杨海玲等[8]研究表明,栀子茵陈中均含有绿原酸,不具备专属性,因此在定性鉴别和定量检测中没有进行绿原酸的检测。
首先参照了《中国兽药典》2015年版第二部中栀子药材的薄层色谱方法,以乙酸乙酯-丙酮-甲酸-水(5∶5∶1∶1)作为展开剂,再喷以100 mL/L硫酸乙醇溶液105℃加热显色,结果加味茵陈蒿汤样品溶液中斑点清楚,但阴性对照有干扰。然后参照赵颖等[9]在清火抗毒丸中栀子薄层色谱法样品处理方法,并考察了以下展开剂:乙酸乙酯-丙酮-甲酸-水(5∶5∶1∶1)(Ⅰ)[10],三氯甲烷-甲醇-冰醋酸-水(10∶4∶1∶1)(Ⅱ),乙酸乙酯-丙酮-水(5∶5∶0.6)(Ⅲ),三氯甲烷-甲醇(3∶1)(Ⅳ)[11]。使用展开剂(Ⅰ)和(Ⅱ),斑点清晰,但阴性有干扰。使用展开剂(Ⅳ)样品分离不开,且有拖尾。使用展开剂(Ⅲ)后,样品斑点较圆且清晰,阴性对照无干扰。所以选择乙酸乙酯-丙酮-水(5∶5∶0.6)为加味茵陈蒿口服液中栀子苷展开剂。

表2 口服液中大黄素加样回收率试验Table 2 Experimental results of the recovery rate of emodin in oral liquid
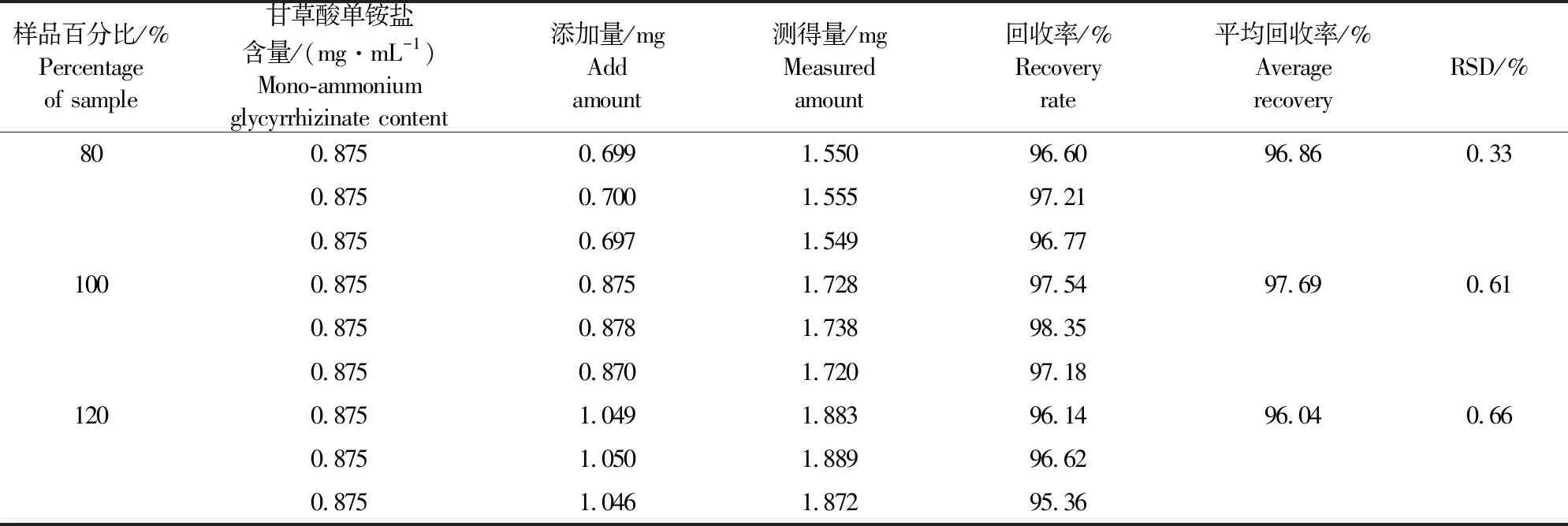
表3 口服液中甘草酸单铵盐加样回收率试验结果Table 3 Experimental results of the recovery rate of Mono-ammonium glycyrrhizinate in oral liquid
大黄素的薄层鉴别,考察了以下展开剂:石油醚(30~60℃)-甲酸乙酯-甲酸(15∶5∶1)的上层溶液(Ⅰ)和正己烷-乙酸乙酯-甲酸(30∶10∶0.5)(Ⅱ)[12]。使用展开剂(Ⅰ),拖尾严重,斑点不清晰。使用展开剂(Ⅱ),斑点圆且显色清晰,分离度较好,阴性对照无干扰。所以选择正己烷-乙酸乙酯-甲酸(30∶10∶0.5)为加味茵陈蒿口服液中大黄素的展开剂。
甘草酸单铵盐的薄层鉴别,考察了以下展开剂:乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)(Ⅰ),氯仿-乙酸乙酯-甲醇-水(15∶40∶22∶10)下层溶液(Ⅱ)[13],正丁醇-冰醋酸-水(6∶1∶3)(Ⅲ)[14]。使用展开剂(Ⅰ),斑点不清晰且阴性有干扰。使用展开剂(Ⅱ),层析不上去。使用展开剂(Ⅲ),效果较好,所以选择正丁醇-冰醋酸-水(6∶1∶3)为加味茵陈蒿口服液中甘草酸单铵盐的展开剂。
栀子苷的含量测定,考察了以下流动相:乙腈-水(15∶85)(Ⅰ)和乙腈-1 mL/L磷酸水(15∶85)(Ⅱ)。使用流动相(Ⅰ),分离度不好,供试品图中有杂峰干扰。使用流动相(Ⅱ),分离度好,阴性对照无干扰。所以选择乙腈-1 mL/L磷酸水(15∶85)为加味茵陈蒿口服液中栀子苷的流动相。
甘草酸单铵盐的含量测定,参照了《中国兽药典》2015年版第二部中甘草药材的含量测定方法进行供试品溶液处理,考察了以下色谱条件,流动相均为乙腈(A)-1 mL/L磷酸水(B)梯度洗脱(0~8 min,19% A;8~35 min,50% A;35~36 min,100% A;36~40 min,19% A);波长237 nm(Ⅰ)[1];梯度洗脱(0~10 min,20% A;10~60 min,45% A);波长265 nm(Ⅱ)[15];乙腈-1 mL/L磷酸水(37∶63),波长250 nm。使用色谱条件(Ⅰ),甘草酸单铵盐对照品不出峰。使用流动相(Ⅱ),基线不稳,杂峰多。使用流动相(Ⅱ),分离度高,阴性对照无干扰。所以选择乙腈-1 mL/L磷酸水(37∶63),检测波长为250 nm作为加味茵陈蒿口服液中甘草酸单铵盐的色谱条件。
结果表明,上述试验方法有效且可靠,可作为加味茵陈蒿口服液的质量控制标准。
猜你喜欢
世界科学技术-中医药现代化(2021年5期)2021-11-05 06:55:48
Journal of Traditional Chinese Medicine(2021年6期)2021-08-09 13:27:22
中成药(2021年5期)2021-07-21 08:38:16
中国化工贸易·中旬刊(2019年10期)2019-10-21 17:46:46
中成药(2017年12期)2018-01-19 02:06:57
广州大学学报(自然科学版)(2015年4期)2015-12-23 11:50:10
丝绸(2015年11期)2015-02-28 14:56:49
天然产物研究与开发(2014年3期)2014-04-27 14:15:32
中国洗涤用品工业(2012年4期)2012-03-20 15:39:34
化学分析计量(2011年1期)2011-04-11 13:13:24
