
Fe、Ir掺杂单层MoS2的构型及电子结构的第一性原理研究
2022-04-06 12:02肖香珍银召利张建伟胡林峰
河南科技学院学报(自然科学版) 2022年2期
肖香珍,银召利,张建伟,胡林峰
(1.河南科技学院实验管理中心,河南 新乡 453003;2.新乡工程学院信息工程学院,河南 新乡 453700)
单层MoS2是具有与石墨烯类似层状结构的过渡金属硫化物,但又与石墨烯不同,常表现出特殊的热力学、光学、电学、磁学等物理化学性质[1-3].近年来,单层MoS2在催化、气体传感、晶体管等领域被广泛应用[4],在某些情况下已成为辅助石墨烯甚至替代石墨烯的重要材料之一.但单层MoS2表面表现出催化惰性[5],整体性能受限于活性位点的劣势.目前,替换式掺杂和吸附式掺杂均是改进材料表面活性的最有效的方法[6-7].Komsa等[8]通过透射电子显微镜观察到过渡金属原子掺杂到单层MoS2表面的过程,结果显示掺杂之后单层MoS2表面活性得到极大提高.Lin等[9]研究了不同掺杂原子对表面磁矩和电子结构的影响,与本征MoS2表面相比,掺杂Mn、Fe、Co原子之后表面磁矩发生了改变.Zhu等[10]将过渡金属分别掺杂MoS2,计算了相应掺杂表面结构稳定性、电子结构,结果显示掺杂体系均表现出高的吸附性能和化学活性.本文对Fe、Ir替换和吸附单层MoS2两种掺杂方式的几何结构、电子结构等进行了对比研究,以期得到不同金属原子对改进MoS2催化活性的意义.
1 计算结构和方法
单层MoS2晶体结构类似于石墨烯[11],具有二维层状结构,三个原子层中的原子呈紧密的S-Mo-S夹心面包式排列,层内原子以共价键结合[12].本研究中MoS2表面掺杂和吸附Fe、Ir的结构均采用Slab超胞模型模拟,所建模型选取3×3×1周期性单层MoS2超胞(Slab模型如图1-C所示),由27个原子组成,考虑到周期性计算可能引起超晶胞层之间的相互作用,平板间真空层厚度设置为1.500 nm,保证层间的作用不受影响.单层MoS2超胞的俯视图、侧视图及Slab模型如图1所示.

图1 单层M oS2超胞的俯视图、侧视图及Slab模型Fig.1 Top view,side view and slab model of single layer MoS2 supercell
图1-A、图1-B分别是周期表面模型的俯视图和侧视图,俯视结构呈平面六角阵列方式排列,Mo原子层与上下S原子层的距离为0.650 nm.原子间距结构优化后本征单层MoS2的晶胞参数为0.319 nm,与实验值0.316 nm接近[13].单层MoS2厚度为0.314 nm,键角∠Mo-S-Mo为81.8°,S-Mo键的键长为0.240 nm.
本研究基于赝势平面波基组的密度泛函理论(DFT)方法[14-15],采用VASP软件包[16-17],离子芯势采用PAW方法描述[18],交换关联泛函采用GGA-PW91形式[19],平面波截止能选取为500 eV,不可约布里渊区积分采用Monkhorst Pack方案[20],K-point设置为5×5×1积分网格.在计算优化过程中所有原子位置均放开优化.通过频率分析对所有优化得到的最优构型加以验证,即局域极小点对应全部实频.
2 结果与讨论
2.1 Fe、Ir在Mo、S位置吸附式掺杂单层MoS2的几何结构
为了研究掺杂体系的稳定性,计算了不同吸附位点的吸附能来确定吸附体系X/MoS2的最优构型.对于单原子吸附在单层MoS2上有3种位置可能存在,如图1-A所示,T m代表Mo原子的顶位,T s代表S原子的顶位,fcc代表MoS2六圆环的中心上方.表1列出了吸附式掺杂在完整表面本征单层MoS2各个位置的吸附能.吸附能 Eads定义式如下

式(1)中:Ex+MoS�代表元素吸附在MoS2表面后体系的总能量, EMoS�代表单层MoS2表面的总能量,Ex代表单个原子的总能量.吸附能可以反映吸附前后各物质总能量的变化[21], Eads的值越大,吸附能力就越强.
Ir、Fe吸附在本征单层表面几何构型的俯视图和侧视图如图2所示.不同体系不同位置对应的吸附能Eads及平均键长dMo-S见表1.

图2 单原子Ir、Fe吸附在单层MoS2上三种不同位置的最优结构Fig.2 Theoptimal structures of monatomic Ir and Fe adsorbed on monolayer MoS2 at threedifferent positions
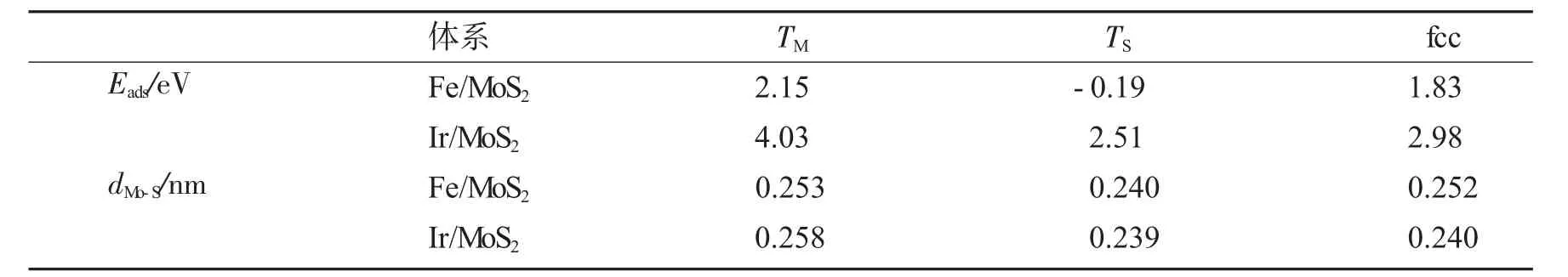
表1 不同体系不同位置对应的吸附能E ads及平均键长d Mo-STable1 Theadsorption energy and average bond length corresponding to different positions of different systems
当Ir、Fe吸附在本征单层MoS2的Mo原子上方时,构型如图2中的A和D.如表1所示,吸附能分别为4.03 eV、2.15 eV,Ir对应下方三个相邻Mo-S键的平均键长为0.258 nm,Fe吸附时对应S-Mo键长平均为0.253 nm,和本征态Mo-S键长0.240 nm相比均被拉长.Ir与邻近的三个S原子之间的距离均为0.222 nm,高于Fe-S键长0.012 nm.
当Ir吸附在S原子上方时,如图2-B、图2-E所示,下方邻近的Mo-S键长平均值是0.239 nm,Fe原子吸附时对应Mo-S键长平均为0.240 nm.Ir、Fe两原子到载体的垂直距离分别为0.208 nm、0.199 nm,相差值为0.090 nm.主要考虑由于Ir的共价半径(0.136 nm)与Fe的共价半径(0.127 nm)之差也为0.090 nm.此时Ir、Fe与邻近的Mo原子的平均键长依次为0.406 nm和0.400 nm.
在fcc位置吸附时,如图2-C、图2-F所示,dIr-S为0.228 nm,dFe-S为0.204 nm.下方邻近的6个Mo-S键长平均值是0.240 nm,Fe原子吸附时对应Mo-S键长平均为0.242 nm.
对比发现两种原子的吸附引入对下方载体的晶格影响较小,说明吸附式掺杂样品的晶体结构没有明显变化.经计算Ir比Fe在表面的吸附性更强,且在Mo原子的上方吸附最强,吸附能为4.03 eV.
2.2 态密度分析
为了进一步分析Fe、Ir原子掺杂单层MoS2对态密度的影响,选取了完整单层超胞MoS2和Ir在Mo位吸附的Mo原子分波态密度,-20~5 eV之间的能量,如图3所示.

图3 本征态及吸附掺杂MoS2的部分态密度Fig.3 Partial density of states of intrinsic states and adsorbed doped MoS2
通过图3-A和图3-B对比分析,可以看出本征态MoS2与吸附MoS2的Mo原子d轨道态密度分布相差较大,吸附MoS2的态密度峰变得平坦且宽,能量主要分布范围在-8.50 eV和3.00 eV之间,本征态的能量主要在-9.00 eV和-0.30 eV.发现吸附Ir原子下方对应Mo原子各个轨道的电子均向高能量方向移动,而Ir原子的5d轨道电子则向低能量方向移动,原因在于Mo原子的4d轨道电子贡献给了吸附Ir原子,Mo原子失去电子,成为施主,体系中Ir原子为受主,当俘获这些电子之后,能量下沉.可见吸附原子与载体单层MoS2之间存在原子轨道的电荷转移和杂化,从而对整个体系的活化起到了关键作用.在此基础上,对比图3-B和图3-C,进一步研究了吸附Ir原子与下方对应Mo原子的哪些轨道存在相互作用,发现在Z方向上,Mo原子的4dyz、4dz2、4dxz轨道与Ir原子的5dyz、5dz2、5dxz态密度峰存在不同程度的混合.特别是dz2轨道之间存在明显混合,且低能量区域-8.30 eV和-2.00 eV之间两个原子都有较高的态密度能,也是两轨道杂化最明显的能量区域,如图3-D.
2.3 Fe、Ir在掺杂缺陷单层MoS2的几何结构
为了获得更优的吸附结构,进一步对单个金属原子掺杂替换本征态表面的S、Mo原子进行了计算.Fe、Ir掺杂替换S、Mo原子的替换能及掺杂原子与相邻原子的平均键长见表2.Ir、Fe替换掺杂的结构如图4所示.

图4 Ir、Fe替换掺杂的结构Fig.4 The structureof Ir and Fesubstitution doping

表2 Fe、Ir掺杂替换S、Mo原子的替换能及掺杂原子与相邻原子的平均键长Tab.2 Thereplacementenergyof Sand Moatomsdoped with Feand Irand averagebond lengthbetweendopedatomand adjacentatoms
Ir、Fe替换S原子掺杂后,dIr-S键的键长为0.239 nm,dFe-S键长为0.228 nm,Mo原子替换后dIr-Mo键长为0.257 nm,dFe-Mo键长为0.240 nm.由于掺杂替换原子半径比S原子半径大,掺杂之后向表面突出,Ir原子的共价半径较大,表现更明显,如图4-A.掺杂之后体系的稳定性由掺杂替换能ΔE来分析

式(2)中:ΔE为掺杂能,其值越小体系越容易掺杂替换,为掺杂体系的总能, EMo/S为 掺杂Mo或S原子, E为单层超胞体系的总能量, Ex为掺杂原子的能量.
根据表2可知,单层MoS2的S位置更容易被替换形成掺杂体系,过渡金属原子Ir相比较比Fe原子更容易形成掺杂体系.试验合成过程中也发现更容易产生S空位缺陷[8].为此S位掺杂体系稳定性强于Mo位掺杂的体系.
2.4 态密度分析
为了详细分析金属替换掺杂对表面体系的影响,本研究对本征MoS2和Ir掺杂替换MoS2的S原子体系进行了态密度计算.本征MoS2的Mo原子及Ir掺杂相邻Mo原子的4d轨道的态密度图如图5所示.
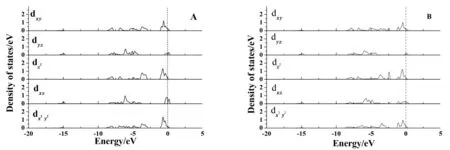
图5 单层超胞Mo原子及Ir掺杂相邻Mo原子的4d轨道态密度Fig.5 The 4d orbital density of statesof Moand adjacent Moatomsdoped with Ir
进一步计算了Ir原子的4d轨道与相邻Mo原子之间的局域态密度,结果如图6所示.

由图6可知,图6-A显示掺杂金属Ir的5dxz和相邻Mo的4dxz在-6.5 eV和-4.5 eV能级附近产生了强烈的杂化作用,说明Ir-Mo之间形成了稳定的共价键,使整个体系具有良好的稳定性.图6-B显示,Ir的和相邻Mo的在能级-4.5 eV和-2.5 eV也有明显的杂化作用.
3 结论
与本征态单层MoS2相比,金属原子的吸附引入均会导致杂质原子附近的MoS2晶格发生畸变,但程度不大,说明两种原子的吸附引入对下方载体的晶体结构影响较小.经计算比较,体系Ir/MoS2稳定性强于Fe/MoS2,Ir更容易吸附在单层MoS2表面,且最优位置在Mo原子的上方,吸附能为4.03 eV,也就是Mo位吸附稳定性强于S位.电子结构态密度分析发现吸附原子Ir与载体单层MoS2之间存在原子轨道的电荷转移和杂化,Ir原子的态密度峰与下方对应Mo原子的轨道与Ir原子的存在不同程度的混合.
Fe、Ir金属原子替换掺杂本征表面MoS2的S、Mo原子的计算结果显示:单层MoS2的S位置更容易被替换形成掺杂体系,S位掺杂体系稳定性强于Mo位掺杂的体系;态密度计算显示,S原子被金属Ir替换后,与Ir邻近的Mo原子的轨道态密度峰较本征态明显变得平且宽,进一步的局域态密度分析显示Ir的5dxz和相邻Mo的4dxz在-6.50 eV和-4.50 eV能级附近产生了强烈的杂化作用形成了稳定的Ir-Mo共价键.
猜你喜欢
分子催化(2022年1期)2022-11-02
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中国建筑金属结构(2018年4期)2018-05-23
科技创新与应用(2017年26期)2017-09-12
中学生数理化·八年级物理人教版(2015年12期)2016-01-25
中学生数理化·八年级物理人教版(2015年12期)2016-01-25
中学生数理化·八年级物理人教版(2015年12期)2016-01-25
