
建兰花叶病毒广东分离物的全基因组序列分析
2022-03-28 13:37魏永路刘伟平朱根发杨凤玺
广东农业科学 2022年2期
魏永路,任 锐,高 洁,刘伟平,谢 琦,朱根发,杨凤玺
(1.广东省农业科学院环境园艺研究所/广东省园林花卉种质创新与利用重点实验室,广东 广州 510640;2.广东翁山兰花研究有限公司,广东 韶关 512600)
【研究意义】建兰花叶病毒(Cymbidium mosaic virus,CymMV)属于马铃薯X 病毒属(Potaxvirus),是分布最广、危害最重的兰花病毒病原之一。CymMV 侵染引起的兰花病毒病在荷兰、美国、韩国、日本和东南亚,以及我国浙江、广东和海南等兰花主产区均广泛流行[1-11]。CymMV的寄主广泛,能侵染兰属(Cymbidium)、卡特兰属(Cattleya)、石斛属(Dendrobium)、蝴蝶兰属(Phalaenopsis)和万代兰属(Vanda)等诸多兰科植物[12-13]。CymMV 侵染兰花后常造成叶片褪绿、枯斑、坏死,植株矮化和畸形等症状,严重影响其生长发育和观赏价值,造成极大的经济损失[14]。目前国际上还没有有效的防治植物病毒病的药剂,但频繁的国际国内贸易、种质资源交换及分株的繁殖方式,均加剧了CymMV等兰花病毒病的广泛传播,对兰花产业化发展构成严重威胁。因此,研究CymMV 全基因组对病毒的检测、监测预警和脱毒技术的开发具有重要意义
【前人研究进展】国内外对兰花病毒病的流行及检测和病毒分子生物学等相关研究已逐步展开。研究发现,CymMV 基因组包含1 条单链正义RNA(约6 200 nt),编码多个功能蛋白,包括依赖RNA的RNA 聚合酶(RNA-dependent RNA polymerase,RdRp)、运动蛋白(Movement Protein,MP)和外壳蛋白(Coat Protein,CP),其中,MP 编码基因包含3 个开放阅读框(TGB1、TGB2 和TGB3)[15]。RdRp 具有介导病毒复制的功能,MP 和CP 蛋白能分别介导病毒细胞间和长距离移动。最新研究表明,当CymMV 和齿兰环斑病毒(Odontoglossum ringspot virus,ORSV) 复合侵染蝴蝶兰时,其小RNA 在感染宿主和免疫逃逸过程中发挥重要作用[16]。对受到CymMV 侵染的国兰进行分析,结果表明,CymMV 侵染确实可以促进国兰1 个非表达病程相关蛋白(Nonexpresserof Pathogenesis Related Genes 1,NPR1)基因和2 个病程相关蛋白(Pathogenesis Related Genes 1,PR1)基因表达[17],基于病毒与宿主的相互作用开发快速检测传感器成为可能[18-19]。对病毒全基因组序列及其功能蛋白编码基因的研究,为病毒关键致病因子的挖掘、病毒-兰花分子互作机制的解析及抗病毒病基因工程等相关研究打下基础[20-24]。
【本研究切入点】目前GenBank 数据库共登录了14 个CymMV 分离物的全基因组序列,其中包括2 个日本分离物Cat(LC125633.1)和Japan(AB197937.1),1个韩国分离物Korean type 2(AF016914.1),1 个印度分离物plm1(AM055720.1),1 个新加坡分离物Singapore(U62963.1),3 个美国分离物Clone 18-29(EF125180.1)、Clone 18-1(EF125178.1)和Clone 18-30(EF125179.1),以及6 个中国分离物NJ-1(JQ860108.1)、China(KR185347.1)、SMi2(AM055640.2)、HNXL(HQ681906.1)、Taiwan(AY571289.1)和M2(EU314803.1)。上述6 个中国分离物分别来自南京、海口、云南和台湾等地,其自然寄主包括海南红壳松(Cephalotaxus hainanensis)和香荚兰(Vanilla planifolia)等。迄今为止未见有关来自广东省CymMV 分离物全基因组序列的报道。【拟解决的关键问题】为了探究兰属CymMV的遗传信息和演化,本研究在对广东兰属花卉主产区病毒调查和检测的基础上,首次进行CymMV 广东分离物病毒全基因组测定、序列同源性分析、系统进化分析和选择压力分析,以进一步明确广东地区CymMV的病毒种类及分类地位,以期为广东省兰花病毒病的监测预警和兰花抗病毒病基因工程提供重要的科学依据和试验材料。
1 材料与方法
1.1 试验材料
CymMV 分离物GZV13 和ZC29 分别来源于广东省广州市天河区和增城区,均从疑似病毒病墨兰叶片中分离纯化,经反转录聚合酶链式反应(RT-PCR)及双抗体夹心酶联免疫吸附分析(DA-SELISA)[25-30]检测均为CymMV 阳性。
总RNA 提取试剂盒和大肠杆菌DH5α 感受态细胞购自天根生化科技(北京)有限公司;cDNA第一链合成试剂盒PrimeScript®1st Strand cDNA Synthesis Kit、DNA 聚合扩增酶PrimeSTAR®Max DNA Polymerase、TA克隆载体pMD19-T Vector、5′RACE 和3′RACE 试剂盒均购自日本TaKaRa Bio 株式会社;DNA 纯化试剂盒和质粒小量提取试剂盒购自美国Omega Bio-Tek 公司;其他化学药品均为国产分析纯。本氏烟种子(Nicotiana benthamiana)由广东省园林花卉种质创新与利用重点实验室保存。
1.2 试验方法
1.2.1 引物合成 从GenBank 数据库中获得已公布的14 个CymMV 全基因组序列(表1),在NCBI 网站上进行序列在线分析。选择相对保守区段,用Primer Premier 5.0 软件[31]设计特异性引物,用于2 个CymMV 分离物的基因组全序列测序(表2)。相邻的两个扩增片段之间至少有800 bp的重叠区域。引物由美国英杰生命技术有限公司合成。
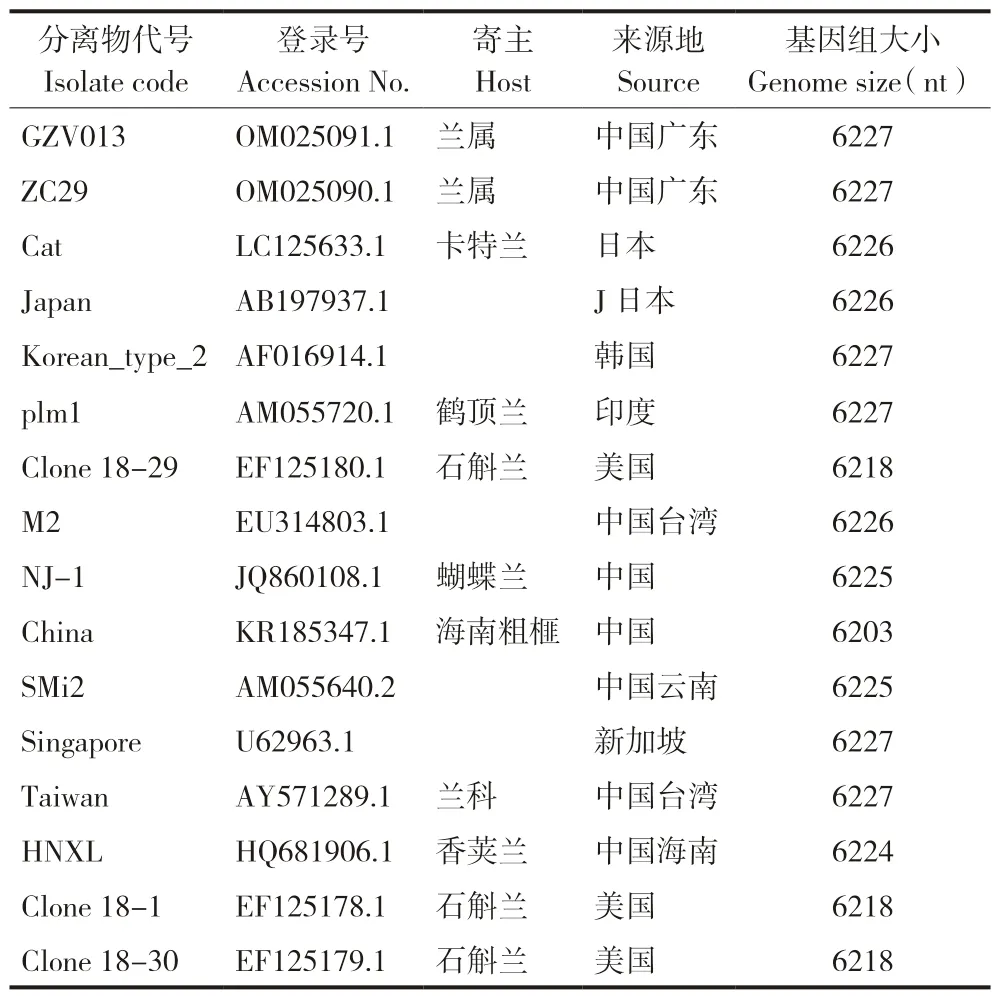
表1 16 种已测序完成的CymMV 全基因组Table 1 16 genomes of CymMV

表2 用于CymMV 病毒测序的引物序列信息Table 2 Information of primers used for the sequencing of CymMV disease
1.2.2 总RNA 提取、cDNA 第一链合成 将CymMV 分离物GZV13 和ZC29 接种于本氏烟20~30 d 后,取发病的新鲜上位叶片,用Total RNA Kit 提取植物总RNA,用Oligo (dT) primers和PrimeScript®1st Strand cDNA Synthesis Kit反转录合成cDNA。
1.2.3 RT-PCR 扩增及凝胶电泳检测 以反转录获得的cDNA 为模板,以引物组合CymMV-N-F/CymMV-N-R、CymMV-M-F/CymMV-M-R 和CymMV-C-F/CymMV-C-R-3 进行RT-PCR 扩增。PCR 反应体系为50 μL,包括10× PCR 缓冲液5 μL,dNTPs(2.5 mmol/L)2 μL,正、反向引物(10 μmol/L)各1 μL,反转录模板cDNA 2 μL,Taq DNA 聚合酶(2.5 U/μL)0.5 μL,补足ddH2O 至50 μL。反应条件为:94℃预变性4 min;94 ℃变性45 s、54~ 68 ℃退火45 s、72 ℃延伸1 min,30 个循环;72 ℃延伸10 min。取5 μL PCR 产物加入2%琼脂糖凝胶进行电泳检测。
1.2.4 CymMV 全基因组序列测定与基因组结构预测 对获得预期大小的病毒基因组片段CymMV-N、CymMV-M 和CymMV-C 进行割胶回收后连接到pMD19-T 载体上,转化大肠杆菌DH5α 感受态细胞。挑取阳性克隆摇菌,对经菌液PCR 检测正确的单克隆提取质粒,进行测序。测序由深圳华大基因股份有限公司完成。序列经Blast 比对,确定为CymMV 基因组片段后,用Contig Express 软件进行CymMV 分离物GZV13 和ZC29的基因组序列拼装,并用NCBI 网站的ORF finder 程序,对CymMV 全基因组序列各功能蛋白编码基因的开放阅读框(Open Reading Frame,ORF) 及基因组结构进行预测,并提交至GenBank数据库。
1.2.5 CymMV 全基因组序列分析、系统进化分析及选择压力分析 对CymMV 全基因组序列及各功能蛋白编码基因,通过在线软件Clustal Omega(https://www.ebi.ac.uk/Tools/msa/clustalo/)进行序列比对和同源性分析;采用MEGA 5.10 软件进行系统进化分析,通过Neighbor-joining 方法构建系统进化树,bootstrap 值为1 000,并隐藏可信度小于70%的值;采用DNASP 5.0 软件进行基因组核苷酸多态性分析及选择压力中性检测,通过计算Pi值估算核苷酸多样性。
2 结果与分析
2.1 CymMV 分离物基因组片段扩增及结构预测
以反转录获得的cDNA 为模板,分别以3对引物进行PCR 扩增,最终获得长度分别为2 800、2 825、3 102 bp的病毒基因组扩增片段CymMV-N、CymMV-M 和CymMV-C(图1)。测序及Blast 序列比对发现,这些片段均为CymMV基因组序列。用Contig Express 软件进行CymMV基因组序列拼装。

图1 CymMV 分离物基因组片段扩增及结构预测Fig.1 Genome fragment amplification and structure prediction of CymMV isolates
除3'-端poly (A) 外,CymMV 分离物GZV13和ZC-29的基因组全长均为6 227 nt,5'-和3'-非翻译区(UTR) 分别为73、76 nt。GZV13 全基因组核苷酸序列中A、G、C 和T 碱基含量分别为26.53%、19.59%、29.05% 和24.83%,ZC-29 全基因组核苷酸序列中A、G、C 和T 碱基含量分别为26.4%、19.83%、29.03% 和24.73%。全基因组编码5 个功能蛋白,包括RdRp(4 254 nt)、TGB 1(701 nt)、TGB 2(339 nt)、TGB 3(276 nt)和CP(276 nt)。以上结果表明,GZV13 和ZC-29 与大部分已报道的CymMV 分离物的基因组结构相同。
2.2 序列一致性分析
为了探寻CymMV 分离物GZV013 和ZC-29与其他已报道分离物之间的差异,对其基因组核苷酸序列和各功能蛋白氨基酸序列进行一致性分析。
全基因组核苷酸序列分析表明,16 个CymMV 分离物中序列一致性最高的是Clone 18-1和Clone 18-30,为98.31%;最低的是HNXL 与Clone 18-30,为86.85%;GZV013 与ZC-29 一致性介于两者之间,为97.01%。与GZV013 和ZC-29 一致率最高的分别是Korean_type_2 和NJ-1,分别为97.36%和97.69%;最低的均是Clone 18-30,均为87.40%。
各区段核苷酸序列分析表明,5'UTR、3'UTR、RdRp、TGB1、TGB2、TGB3 和CP基因的一致率分别为86.11%~100%、82.86%~100%、8 7.0 2%~9 6.6 7%、8 2.8 8%~9 8.5 8%、8 7.0 9%~9 8.5 2%、8 9.4 9%~9 9.2 8% 和89.43%~99.11%。如表3 所示,各分离物中,与GZV013的5'UTR 一致性最高的是Korean type 2(97.36%),最低的是ZC29(91.67%);其3'UTR 与ZC29、China、HNXL 和Korean type 2 完全一致(100%),而其与Taiwan的差异最大(82.86%);与GZV013的RdRp、TGB1、TGB2、TGB3 和CP 基因序列一致性最高的分别 是Taiwan(97.27%)、Japan(97.86%)、Taiwan(98.23%)和Singapore(99.64%),最低的均为Clone 18-30,分别为87.09%、84.45%、89.09%、90.94%和97.92%。
各区段氨基酸序列分析表明,各编码蛋白RdRp、TGB1、TGB2、TGB3 和CP的一致率分别为94.71%~97.81%、84.48%~99.57%、9 4.6 4%~1 0 0.0 0%、8 7.9 1%~1 0 0% 和97.31%~99.10%。如表3 所示,各分离物中,与GZV013的RdRp、TGB1 和TGB2 一致率最高的分别是Taiwan(97.67%)、ZC29(99.57%)和Taiwan(100%),其TGB3 序列与NJ-1 和China完全一致(100%),而与其RdRp、TGB1、TGB2和TGB3 一致率最低的均是Clone 18-30,分别为94.71%、84.48%、94.64%和87.91%。与GZV013的CP 基因氨基酸序列一致性最高的包括ZC29、NJ-1、Cat 和Japan,均为99.1%;氨基酸序列一致性最低的是印度分离物plm1 和美国分离物Clone 18-29,均为97.31%。

表3 GZV013 与其他CymMV 分离物的核苷酸和氨基酸序列相似性Table 3 Nucleotide and amino acid sequence identities between GZV013 and other CymMV isolates
综合以上结果,16 个CymMV 分离物全基因组序列一致性为86.85%~98.31%;GZV013 和ZC-29 与中国(NJ-1、China 和HNXL 等)和韩国(Korean_type_2)等邻近广东省的地区分离物的核苷酸和氨基酸序列一致性较高,而与美国分离物Clone 18-1 和Clone 18-30 最低。
2.3 系统进化分析
用 MEGA5.0 软件,对16 个CymMV 分离物进行基于全基因组序列的系统进化分析。结果(图2)显示,16 个分离物共可聚类为A、B 两个类群。其中,8 个中国分离物(ZC29、GZV013、China、NJ-1、SMi2、HNXL、Taiwan 和M2)、2个日本分离物(Cat 和Japan)、1 个韩国分离物(Korean_type_2)、1 个新加坡分离物(Singapore)、1 个印度分离物(plm1)和1 个美国分离物(Clone 18-29)聚类为A 群;2 个美国分离物(Clone 18-1 和Clone 18-30)聚类为B 群。

图2 CymMV 分离物全基因组核苷酸序列的系统进化分析Fig.2 Phylogenetic analysis of the complete genomic nucleotide sequence of CymMV isolates
A 群又可分为Ⅰ和Ⅱ两个亚群。其中,2 个中国分离物(HNXL和China)、2个日本分离物(Cat和Japan)、1 个韩国分离物(Korean_type_2)、1 个印度分离物(plm1)和1 个美国分离物(Clone 18-29)聚类为亚群Ⅰ,这些分离物的自然寄主包括海南红壳松(Cephalotaxus hainanensis)、香荚兰(Vanilla planifolia)、石斛兰属(Dendrobium)和卡特兰属(Cattleya)等。ZC29 与南京分离物NJ-1的亲缘关系最近,GZV013 与台湾分离物M2的亲缘关系最近,它们与中国云南分离物SMi2聚类成亚群Ⅱ,且这5 个分离物的寄主多为兰属和蝴蝶兰属。结果表明CymMV 多样性种群的分化与寄主种类和地理隔离关系密切。
2.4 基因组多态性和选择压力分析
为了研究CymMV 基因组在自然选择压力下的进化和遗传变异,对16 个CymMV 分离物的全基因组序列进行核苷酸多样性分析和中性检测。
从CymMV 不同区域基于Pi值的多态性图谱(图3)可见,RdRp的C 端、TGB1、TGB2在全基因组中Pi值最高,即变异性最高;而5'UTR、3'UTR、RdRp的N 端、TGB2 和CP的Pi值较低,即多态性较低。
通过Tajima 中性检测的D值估计CymMV 基因组每个区域的变异情况,D值越小,受到负选择的影响越大。结果(图3)显示,整个基因组的D值均为负值,说明整个基因组序列都在不同程度上受到负选择压力。
为进一步确定CymMV 种群是否满足中性进化模型,本研究用Fu 和Li 检验计算CymMV 基因组每个区域的F∗值,F∗值越小,受到负选择的影响越大。结果(图3)显示,各F∗值均为负值,与上述D值一致,表明CymMV 基因组每个区域均受到负选择的影响,并符合中性进化模型;RdRp、TGB1 和TGB2 在全基因组中变异性最高。

图3 CymMV 基因组多态性和选择压力分析Fig.3 CymMV genome polymorphism and selection pressure analysis
3 讨论
兰花一般指兰科(Orchidaceae)植物,是被子植物中最大科之一。其中,国兰是中国十大传统名花之一,是广东省最具优势和高产值的花卉种类之一,在国内外花卉市场上占有重要地位[32]。CymMV 作为分布最广、危害最重的兰花病毒病原之一,能侵染兰属、卡特兰属、石斛兰属、蝴蝶兰属和万代兰属等诸多兰科植物。因此,展开兰花病毒病的病害检测和病毒分子生物学等相关研究,将为兰花病毒病防控体系的建立及兰花产业化的健康发展提供支持。
我国是多数地生兰科植物的起源地[33],国兰(Cymbidium)作为重要的特色花卉,被出口到韩国、日本和东南亚各国[34],而蝴蝶兰等洋兰也日益受到青睐。由于频繁的国际和国内贸易、种质资源交流,以及兰花病毒病检测检疫技术手段的局限性,导致兰花病毒病在兰花主产区和主要消费地区广泛传播。本研究发现,CymMV 广东分离物GZV013 和ZC-29,与我国台湾等其他省份以及韩国等邻近国家和地区分离物的序列一致性及亲缘关系最高,说明它们可能有共同的起源。
病毒进化有一定随机性,但又受选择压力而呈现一定稳定性。在兰花病毒病随寄主材料传播的过程中,CymMV 等病毒逐渐进化出能侵染更多兰科植物的新株系。本研究发现与GZV013 和ZC-29 亲缘关系最近的分离物的寄主均为兰属和蝴蝶兰属,与其亲缘关系较远分离物的寄主则包括海南红壳松、香荚兰、石斛兰属和卡特兰属等兰科植物,说明CymMV 多样性种群的分化与寄主种类关系密切。能侵染其他兰科植物的CymMV新分离物的出现,对兰科植物种质资源保护和利用提出了新挑战。因此,研发操作简单、灵敏度高和成本低廉的兰花病毒病检测技术,建立有效的兰花病毒病防控体系是遏制兰花病毒病广泛传播的关键。
CymMV 在不同的选择压力下,会发生基因组突变和遗传进化。本研究表明,CymMV 基因组每个区域均受到负选择的影响,且RdRp、TGB1和TGB2 在全基因组序列中变异度最高,说明自然选择压力下(如新寄主等)这些基因发生的有益变异更多。但无数据表明这些基因显著地受到了更大的选择压力。因此RdRp、TGB1 和TGB2在CymMV 侵染寄主过程中的作用还需要进一步研究。
4 结论
本研究利用RT-PCR、DA-SELISA 和3 对通用引物,对采自广州地区疑似病毒病症状的墨兰进行鉴定,并完成了GZV13 和ZC29 两个毒株的全基因组组装、功能注释及核苷酸序列分析。通过国内外16 个毒株全基因组系统进化分析,明确了广东地区发生的CymMV的分类地位。同时序列相似性也暗示了危害广东兰科植物的CymMV可能来自南京、台湾等地,也可能来自与广东农产品贸易频繁的日本、韩国等国家。提示应在国内地区间及进出口贸易中加强相关检疫措施。基因组多态性和选择压力分析表明,CymMV 易产生大量的低频突变,为病害在兰科植物间的传播带来较大不确定性,提示应在广东地区兰科种植早期做好病虫害综合防治、关键抗病基因筛选及鉴定、抗病品种筛选及靶标药剂研发等方面工作。
猜你喜欢
汽车实用技术(2022年19期)2022-10-19
计算技术与自动化(2022年2期)2022-07-04
汽车实用技术(2022年9期)2022-05-20
当代水产(2022年1期)2022-04-26
华东师范大学学报(自然科学版)(2019年5期)2019-11-11
医学信息(2019年4期)2019-10-08
中国瓜菜(2019年8期)2019-09-19
烹调知识(2019年3期)2019-03-01
江苏农业科学(2016年11期)2017-03-21
江苏农业科学(2017年1期)2017-02-27
