
超声辅助酶法提取-液相色谱-原子荧光光谱法测定富硒黑木耳中硒形态
2022-03-23 07:58刘笑笑张振都宋志峰魏春雁
中国无机分析化学 2022年1期
刘笑笑 金 秋 张振都 张 奇 宋志峰 魏春雁*
(1.吉林省农业科学院/吉林省农业环境与农产品安全重点实验室,长春 130033; 2.吉林省吉测检测技术有限公司,长春 130117)
硒是人和许多生物所必需的微量元素,具有抗氧化、提高免疫力、降低血糖等多种功能[1-7],但其在体内的安全含量存在一个较小的范围,缺少会引发许多疾病,过量亦会引起生物体中毒。
硒元素有益和有毒主要取决于该元素存在的化学形态和含量[8],因此,建立灵敏度高、稳定性好、准确可靠的硒形态检测方法尤为重要。
目前,采用电感耦合等离子质谱、电感耦合等离子体原子发射光谱、原子吸收光谱和原子荧光光谱等元素分析仪结合水提、酸提、酶提等提取方法进行硒形态定性定量分析[9-12],但在实际检测分析时,常面临基质复杂、形态多样和含量低等问题。基于此,采用超声辅助酶法提取-原子荧光光谱法测定,系统分析了影响硒形态提取及分离的因素,建立了适宜富硒黑木耳中硒形态分析检测的方法,为客观评价富硒黑木耳质量提供科学依据。
1 实验部分
1.1 材料与试剂
富硒木耳,采购于伊春金瑞森林食品有限公司,粉碎后,过150 μm筛网,置于阴凉干燥处保存。
硒代蛋氨酸溶液(Selenomethionine,Se-Met,97.9 μg/g)、硒代胱氨酸溶液(Selenocystine,Se-Cys,93.5 μg/g)、甲基-硒代半胱氨酸溶液(Methyl Selenocysteine,Se-MSeC,96.6 μg/g)、亚硒酸根溶液(SeO32-,68.9 μg/g)、硒酸根溶液(SeO42-,75.1 μg/g)均购自中国计量科学研究院,木瓜蛋白酶、链霉蛋白酶E购自Solarbio 生命科学公司,甲醇(色谱纯,赛默飞世尔科技有限公司),磷酸氢二胺(优级纯,天津市光复精细化工研究所),四丁基溴化铵(分析纯,麦克林),盐酸(优级纯,西陇化工股份有限公司),氢氧化钾(优级纯,天津市光复精细化工研究所),硼氢化钾(分析纯,天津市光复精细化工研究所),碘化钾(分析纯,天津市福晨化学试剂厂),甲酸(分析纯,天津市光复精细化工研究所),实验用水均为实验室超纯水。
1.2 仪器设备
SA-10原子荧光形态分析仪(北京吉天仪器有限公司),硒原子荧光空心阴极灯(北京有色金属研究总院),HZQ-C空气振荡器(哈尔滨市东联电子技术),KQ-500DE数控超声波清洗器(昆山市超声仪器有限公司),BT 124S 电子天平(赛多利斯科学仪器(北京)有限公司)。
1.3 实验方法
1.3.1 仪器条件
1)色谱条件
Hamilton PRP-X100离子交换色谱柱(250 mm×4.1 mm,10 μm),流动相为磷酸氢二铵(40 mmol/L)+四丁基溴化铵(0.5 mmol/L),用甲酸(20%)调节pH 值为6.0。流量1.0 mL/min,进样体积100 μL。
2)原子荧光光谱条件
负高压300 V,灯电流90 mA,载气流量400 mL/min,屏蔽气流量600 mL/min,原子化器高度8 mm。
3)氢化物发生条件
碘化钾(4 g/L) + 氢氧化钾(3.5 g/L,氧化剂),硼氢化钾(20 g/L) + 氢氧化钾(3.5 g/L,还原剂),盐酸(10%,载流),转速80 r/min。
1.3.2 标准溶液配制
准确移取各标准溶液2 mL,分别置于50 mL容量瓶中,用超纯水定容至刻度,即得硒酸根(SeO42-,1.660 μg/mL)、亚硒酸根(SeO32-,1.716 μg/mL)、硒代胱氨酸(Se-Cys,1.768 μg/mL)、硒代半胱氨酸(MSeC,1.392 μg/mL)、硒代蛋氨酸(Se-Met,1.576 μg/mL)的标准溶液,4 ℃储存。
分别再用标准溶液配制成40、80、120、160、200 μg/L的混合标准溶液。
1.3.3 待测溶液制备
准确称取样品0.1 g(精确至0.000 1 g)于离心管中,按料液比(1∶60)加入木瓜蛋白酶溶液6 mL,于超声波清洗器中,水温设定37 ℃(链酶蛋白酶E在37 ℃时效率最稳定),提取45 min,每隔10 min摇晃一次离心管。8 000 r/min离心20 min,上清液经0.45 μm微孔水系滤膜过滤,得样品待测液,备上机检测。同时做试剂空白实验。
2 结果与分析
2.1 提取剂的筛选
以普通木耳为试验样品,对5种硒形态分别添加回收,考察了甲醇溶液(50%)、柠檬酸溶液(0.1 mol/L)、氢氧化钠溶液(0.1 mol/L)、木瓜蛋白酶溶液(100 g/L)和链酶蛋白酶 E溶液(3 mg/mL)对5种硒形态的提取效果。
从表1中可以看出,甲醇溶液(50%)、柠檬酸溶液(0.1 mol/L)可以提取亚硒酸和硒酸2种无机硒形态,对于其他3种有机硒形态无法提取。通过亚硒酸的回收率结果分析,可能存在硒形态之间的转化,使亚硒酸的回收率偏高。氢氧化钠溶液(0.1 mol/L)在提取过程中,木耳上清液更加黏稠、浑浊,无法通过滤膜,达不到上清液澄清的提取效果。木瓜蛋白酶与链酶蛋白酶 E 属于蛋白水解酶,其中木瓜蛋白酶对木耳中甲基-硒代半胱氨酸和硒代蛋氨酸提取效果较好,而其他3种形态无法提取;链酶蛋白酶 E 对5种硒形态的提取率高,上清液澄清。
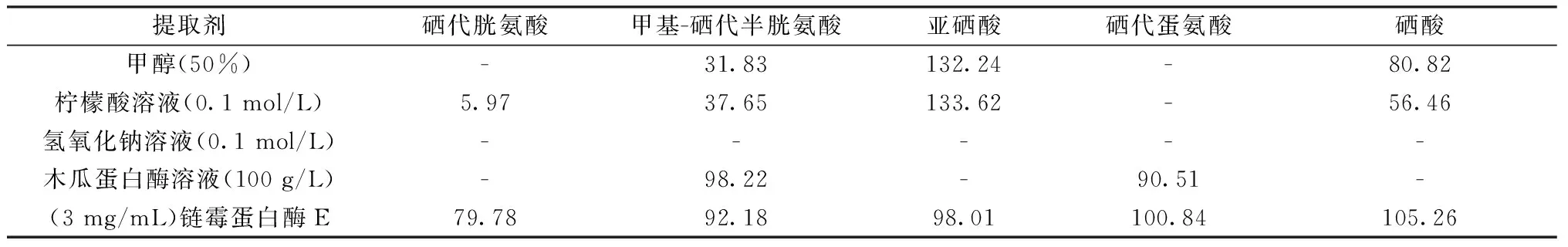
表1 不同提取剂对硒形态提取率的影响
2.2 提取方式、时间及料液比的筛选
木耳多糖含量高,遇水膨胀,上清液黏稠,且黏稠的溶液易将未泡发的木耳粉末包裹住形成结块,附着在容器壁上,泡发不充分,在提取过程中会造成浪费、提取不完全等情况(见图1a)。
从木耳泡发状态上比较,振荡提取方式比超声提取方式更容易使木耳充分的泡发,在振荡过程中,木耳粉末容易散开不形成结块(见图1b)。但是振荡提取时间比超声提取时间长,有机形态通常不稳定,尤其是硒代胱氨酸与硒代蛋氨酸之间容易转化,长时间振荡过程中容易发生分解或转化。因此采用超声辅导提取方式,时间控制在 60 min 以内,5种形态可以保持稳定。在超声的过程中,每间隔 10 min,摇晃一次离心管,防止木耳粉末长时间沉淀在离心管下层结块或附着在管壁上。
从料液比的选择上,分别对1/10、1/20、1/40、1/60、1/80、1/100进行比较。称取木耳0.2 g依次加入纯水2、4、8、12、16 mL,每隔15 min,观察木耳泡发状态。过少的提取液致使木耳遇水膨胀,如图1E和1F所示,1/10和1/20的料液比时,木耳无法泡发完全,有明显的结块,上清液分离不完全,硒元素形态提取不充分,而使用过多的提取液导致最终硒元素形态含量检测较少,结果不准确。1/40、1/60、1/80在30 min内能将木耳泡发完全(见图1c)。1/40在离心过后,上清液过少,不易过膜,经过多次实验确定采取1/60的料液比效果较好(见图1d)。
综上,提取方式上,超声提取优于振荡提取,提取时间60 min为宜,料液比1/60效果相对较好。
2.3 碘化钾浓度的筛选
碘化钾的作用是氧化剂,其与硒酸发生氧化还原反应,将硒酸还原为亚硒酸。碘化钾的浓度直接影响硒酸的荧光强度。分别考察碘化钾质量浓度为3、4、5、6 g/L时5种硒形态的荧光强度。从图 2中可以看出,当碘化钾的质量浓度增加时,只有硒酸和亚硒酸的荧光强度会随着碘化钾浓度的增加而升高,其他硒形态荧光强度差异不显著。而当碘化钾的质量浓度大于 4 g/L时,荧光强度达到最大值。因此选择碘化钾浓度为4 g/L。

图1 木耳中硒形态提取方式、时间和料液比的选择Figure 1 Selection of extracting method,time and ratio of solid to liquid of selenium from auricularia auricula.

图2 碘化钾的浓度对 5 种 硒形态荧光强度的影响Figure 2 Effect of concentration of potassium iodide on fluorescence intensity of five selenium forms.
2.4 流动相浓度的确定
以磷酸盐缓冲溶液为流动相,分别考察了磷酸氢二铵的浓度为30、40、50 mmol/L 时5种硒元素形态的分离情况,结果如表2所示。流动相的浓度对亚硒酸与硒酸出峰时间影响较大,对硒代胱氨酸、甲基-硒代半胱氨酸和硒代蛋氨酸出峰时间无明显变化。随着流动相浓度升高,亚硒酸与硒酸出峰时间均提前。当流动相浓度为30 mmol/L 时,5种硒形态在 30 min 全部出峰,而亚硒酸与硒代蛋氨酸有重叠。当流动相浓度为40 mmol/L 时,亚硒酸与硒代蛋氨酸全部分离,5种硒形态在16 min 内全部出峰。当流动相浓度为50 mmol/L 时,5种硒形态全部出峰时间缩短在12 min 内,但亚硒酸又与前面的甲基-硒代半胱氨酸峰有重叠。因此,在5种硒形态峰完全分离的条件下,流动相浓度40 mmol/L 为最佳检测条件(图3)。

表2 不同流动相浓度对测定结果的影响

图3 5种硒形态流出曲线图Figure 3 The peak time five selenium forms.
2.5 线性关系实验
准确移取5种硒形态混合标准溶液浓度为40、80、120、160、200 μg/mL,用上述仪器条件依次测定,5种硒元素形态在16 min内全部出峰,顺序依次为硒代胱氨酸、甲基硒代-半胱氨酸、亚硒酸、硒代蛋氨酸、硒酸。
从图4中可以看出,硒代胱氨酸、硒代半胱氨酸、亚硒酸、硒蛋氨酸、硒酸标准曲线的相关系数分别为0.999 0、0.999 2、0.999 9、0.999 4、0.999 9。
2.6 检出限实验
按照上述仪器条件,以试剂空白溶液为测试基准,连续测定11 次,计算空白值的标准偏差,DL=3SD/K(DL为检出限,K为校正曲线斜率);5种硒元素形态检出限依次为:硒代胱氨酸0.35 μg/L,甲基硒代半胱氨酸 0.46 μg/L,亚硒酸 0.26 μg/L,硒代蛋氨酸 0.64 μg/L,硒酸 3.06 μg/L。
2.7 精密度实验
连续测定相同浓度标准溶液6针,计算各组分峰面积的相对标准偏差(RSD)。5种硒元素形态RSD为别为2.4%、1.9%、0.90%、0.91%、2.0%。参见表3。
2.8 稳定性实验
同一份样品溶液(溶液中5种硒形态本底值约为硒代胱氨酸60 μg/L、甲基硒代胱氨酸120 μg/L、亚硒酸120 μg/L、硒代蛋氨酸180 μg/L、硒酸120 μg/L)分别在0 、2、4、8、12 h时上机测定,测定结果计算RSD。5种硒元素形态RSD依次为1.7%、2.2%、2.7%、1.7%、1.6%,见表4。
2.9 加标回收实验
取已知硒形态含量的富硒木耳6份,分别添加适量5种硒形态浓度,按上述样品制备并定容至5 mL,使得添加浓度为30、60、120 μg/L(已知木耳中硒代蛋氨酸上机液浓度约为60 μg/L),上机测定并计算回收率及RSD。结果表明,5种硒元素形态的加标回收率在76.1%~108%,RSD值均小于3.9%,结果见表5。

图4 5种硒形态标准曲线图Figure 4 5 Selenium form standard curve.

表3 精密度考察结果
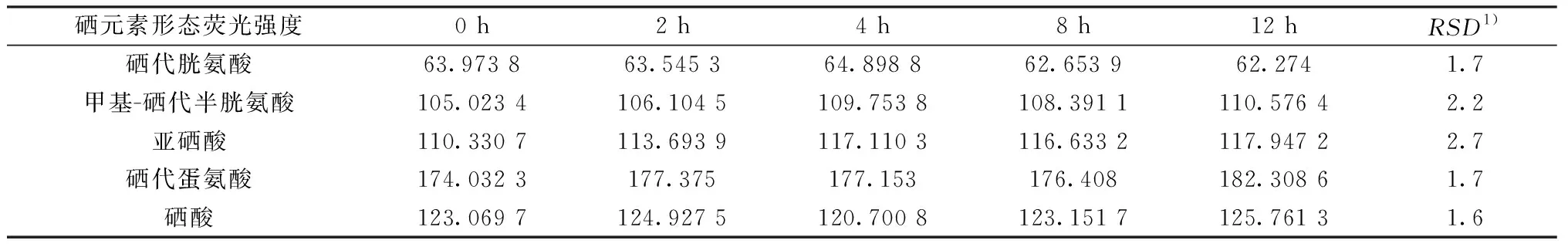
表4 稳定性考察结果
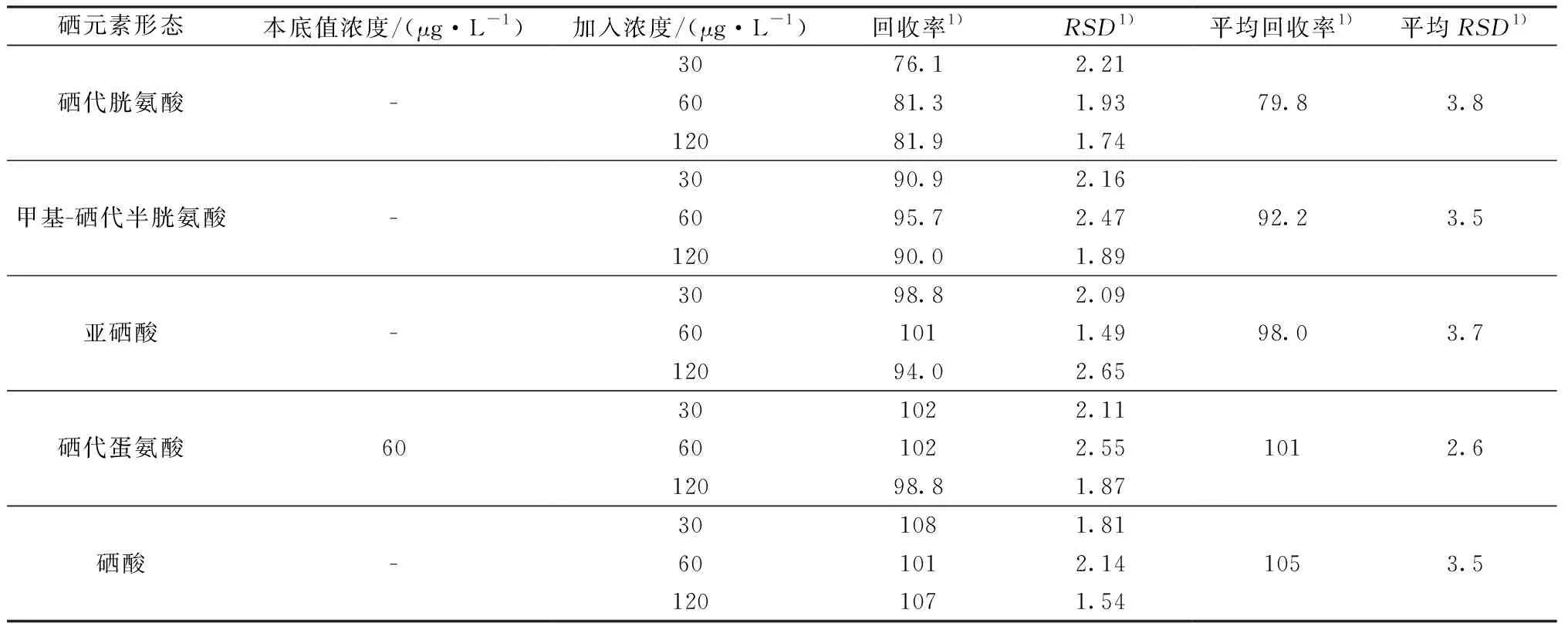
表5 加标回收实验
3 结论
1)测定富硒木耳中硒形态含量时提取剂为链酶蛋白酶E,方式为超声提取,提取时间为60 min,料液比为1/60。流动相磷酸氢二铵为40 mmol/L,碘化钾为4 g/L。
2)测定富硒木耳中硒形态含量时5种硒形态硒代胱氨酸、硒代半胱氨酸、亚硒酸、硒蛋氨酸、硒酸标准曲线的相关系数达到0.999 0、0.999 2、0.999 9、0.999 4、0.999 9,检出限低,分别为硒代胱氨酸0.35 μg/L、甲基-硒代半胱氨酸 0.46 μg/L、亚硒酸 0.26 μg/L、硒代蛋氨酸 0.64 μg/L、硒酸3.06 μg/L,加标回收率为 76.1%~108%,精密度高、重现性好,方法稳定、准确可靠。
猜你喜欢
生物信息学(2022年3期)2022-11-12
中国饲料(2022年5期)2022-04-26
西南农业学报(2021年10期)2021-12-14
今日农业(2021年10期)2021-07-28
昆明医科大学学报(2021年1期)2021-02-07
猪业科学(2020年12期)2021-01-09
中华养生保健(2020年5期)2020-11-16
中华养生保健(2020年4期)2020-11-16
女士(2017年10期)2017-11-01
作文大王·低年级(2017年2期)2017-02-16
