
数据驱动的机器学习在电化学储能材料研究中的应用
2022-03-23 08:46施思齐涂章伟邹欣欣孙拾雨杨正伟
储能科学与技术 2022年3期
施思齐,涂章伟,邹欣欣,孙拾雨,杨正伟,刘 悦,4
(1上海大学材料科学与工程学院;2上海大学材料基因组工程研究院;3上海大学计算机工程与科学学院;4上海市智能计算系统工程技术研究中心,上海 200444;5之江实验室,浙江 杭州 311100)
作为能源互联网的关键环节,以锂/钠离子电池为代表的储能电池正处在与信息产业深度融合的阶段,目前的发展目标是突破储能电池能量密度低、电池安全性差、大电流充放电能力不足以及使用寿命短等方面的瓶颈并进一步拓宽其应用场景[1]。然而,储能电池的综合性能受各类材料的复杂构效关系共同影响,如电极脱嵌锂过程的结构演化[2]、电解质的离子输运机制[3]和电极与电解质间的界面性质[4]等,这为储能电池的研发和性能提升带来了挑战。
早期的储能电池研发是基于经验主义的实验方法,涉及人工合成、材料表征和性能分析等步骤,耗时长且成本高。于是,研究人员进一步发展了基于物理化学定律的理论研究方法[5],但该方法在解决许多电化学储能材料科学问题时往往过于复杂,难以求解。后来,随着材料科学、物理学和计算机科学的交叉与融合,微观-介观-宏观尺度的计算模拟方法逐渐兴起,包括第一性原理计算、分子动力学模拟、蒙特卡罗模拟、CALPHAD方法、相场模拟和有限元模拟等[6]。这些方法涵盖了不同的空间和时间尺度范围,在可充电电池领域得到了广泛的应用[7-8]。然而,计算模拟方法依赖于材料的微观结构和高性能计算设备,其计算速度和准确性仍然受到限制,且该方法每次往往只能对材料的单一性能进行研究与优化,很难同时筛选或设计出综合性能优异的电化学储能材料。近几年,随着实验、理论和计算数据的大量积累以及高效、准确的人工智能技术的迅速发展,材料科学研究进入了第四科学范式[9-10],即数据驱动的材料科学研究,有望实现储能电池的高效研发。
如图1所示,数据驱动的材料科学利用传统实验、理论和计算模拟方法积累的大量数据,借助数据驱动的人工智能方法对电化学储能材料的性能驱动机制进行建模和分析,以加速新型高性能电化学储能材料的研发与设计。目前,作为数据驱动的人工智能方法的典型代表之一,机器学习已经被广泛应用于材料的性能预测和新材料发现[11-18]。机器学习在电池领域的应用可以追溯到1999 年Salkind等[19]使用模糊逻辑方法来确定电池的充电状态和健康状态。随后,Ceder 等[20-22]利用机器学习技术预测材料晶体结构并用于汽车电池锂基材料的发现。2011 年,美国政府提出了“材料基因组计划”(materials genome initiative,MGI)[23],其目标之一便是通过机器学习方法将“实验”、“计算”和“数据”相结合,以快速开发出清洁能源系统的相关材料[24-25]。自此,以数据驱动的机器学习方法助力电化学储能材料研发的工作不断涌现出来。已有一些优秀综述从不同的角度介绍了电化学储能材料领域中机器学习的研究现状。例如,Guo等[26]从材料原子建模的角度,介绍了机器学习在固态电池材料的势能函数构建、性能预测和逆向设计中的应用;陈翔等[27]从多尺度电池应用的角度,评述了机器学习与微观、介观和宏观尺度的理论或实验融合的方法在电池材料的研究现状;Lombardo 等[28]从材料研发到电池实际应用的角度,总结了机器学习在电池制造、材料表征和电池诊断等方面的研究进展;刘悦等[29]从机器学习工作流程的角度,综述了机器学习在充电电池材料领域的应用现状,并分析和总结了机器学习方法在材料领域应用普遍面临的三大挑战问题和相应的解决策略。
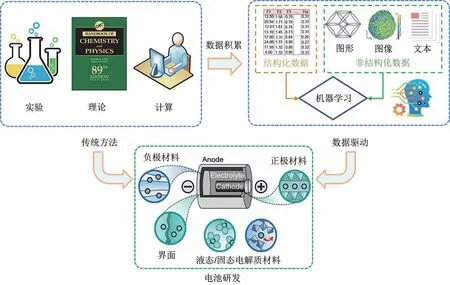
图1 电池研发四大范式:实验、理论、计算和数据驱动Fig.1 Four paradigms of battery research and development:Experimentation,theory,computation and data driven
在MGI的推动下,电化学储能材料数据被不断产生和积累,包括结构化数据和非结构化数据。其中,结构化数据一般能够形式化存储在数据表格中,且每列都有具体的含义;非结构化数据则通常指结构化数据之外的一切数据,包括节点和边组成的图形数据、像素点组成的图像数据和字符组成的文本数据。然而,利用这些异构数据来驱动电化学储能材料的研发,对机器学习建模过程中的数据表示、模型选择、评估与应用提出了新的挑战。本文以不同类型数据驱动的机器学习在电化学储能材料研发中的应用为主线,全面介绍了可用于电化学储能材料研究的材料数据资源,并指出了其未来发展方向;重点总结了结构化数据驱动下机器学习的工作流程及其在电极和电解质材料的性能预测与成分优化、电池健康状态评估的应用现状,以及非结构化数据驱动下机器学习在材料性能预测、表征图像分析和文献文本挖掘等方面的相关工作;系统厘清了机器学习在电化学储能材料领域应用所面临的三大矛盾,并结合机器学习的最新发展提出了相应的调和策略;最后,对全文内容进行了总结。
1 电化学储能材料的数据资源
过去若干年里,全世界范围内材料研究学者们通过实验测量和计算模拟积累了大量的材料数据,由此建立了大量可用于电化学储能材料研究的涵盖材料结构与性能的数据库(表1)。电化学储能材料中重要的性能如脱/嵌锂电位、热力学稳定性和化学稳定性等均可从密度泛函计算得到的能量、电子结构等信息中获得,因此包含这些信息的通用型材料数据库都可用于电化学储能材料本征性质的研究[30]。从这些材料数据库中能够得到电化学储能材料的实验或计算的原始数据,为数据驱动的机器学习提供样本。
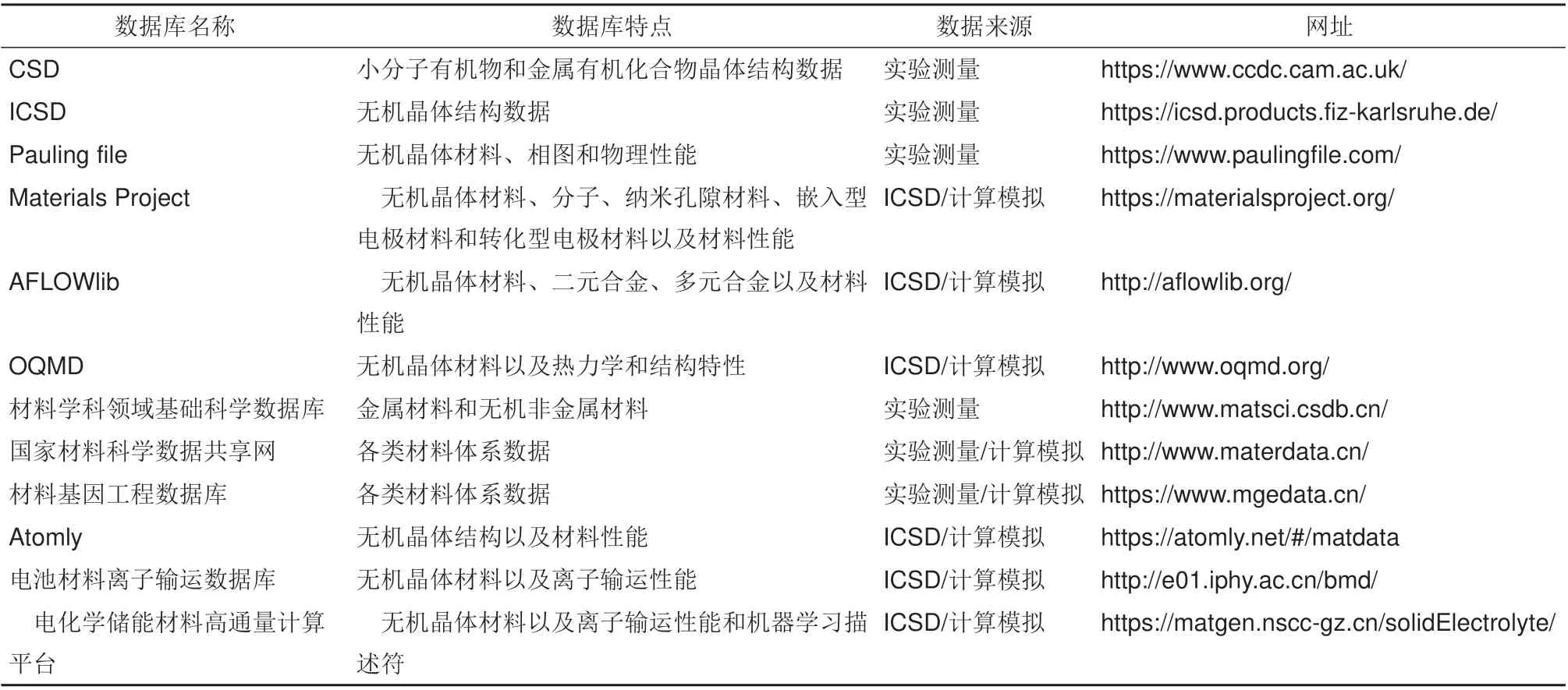
表1 主要的材料数据库及其数据特点Table 1 Main material databases and data characteristics
实验测量作为沿用至今的材料科学研究关键手段之一,对材料的研发起着至关重要的作用。科学工作者们通过对文献中实验测量数据的收集,建立了一些材料数据库,其中包含了化学组成、材料结构、文献引用等基本信息。剑桥结构数据库(cambridge structural database,CSD)由英国剑桥大学Kennard 等在1965 年创建,从文献中收录了115万种小分子有机物和金属有机化合物晶体结构数据,其中包含了晶胞参数、原子坐标和引用文献等[31-33]。德国波恩大学Bergerhoff 等[34]在1983年创建了无机晶体结构数据库(inorganic crystal structure database,ICSD)来作为剑桥结构数据库的补充,收录了1913年以来出版的21万多条实验表征的无机晶体结构详细信息,包含化学名称、化学式、矿物名、晶胞参数、空间群、原子坐标、原子占位及文献引用等[35]。1995年,日本科学技术厅等[36]单位合作组建了Paulina Film 项目,收集了从1900 年至今超过35000 种出版物中的无机材料数据,包含了35 万个晶体结构、5 万个相图和15 万条物理性能。为了有效地应用和积累科学数据,我国在1987 年由中国科学院牵头正式启动科学数据资源建设。其中,中国科学院金属研究所承建的“材料学科领域基础科学数据库”,(http://www.matsci.csdb.cn/)拥有金属材料数据6万余条和无机非金属材料数据1万余条,涵盖了材料的热学、力学和电学等各种性能。2001 年我国开始逐步启动科学数据共享工程,其中北京科技大学建设的“国家材料科学数据共享网”(http://www.materdata.cn/)汇集了全国30余家科研单位包括有色金属材料、有机高分子材料和能源材料等超过60万条材料科学数据。虽然这些基于实验测量的材料数据库记录的数据可靠且直观,但是获得这些数据的成本高昂。
随着计算机算力的提升,材料研究模式开始以“经验试错法”到基于“材料基因”设计方法转变,期间催生了许多高通量材料计算平台和数据库。劳伦斯伯克利国家实验室Ceder 等[37]在2011 年创立Materials Project 数据库,存储了75 万多种材料,涉及无机化合物、分子、纳米孔隙材料、嵌入型电极材料和转化型电极材料以及包括9万多条能带结构、弹性张量、压电张量等性能的第一性原理计算数据。2012 年,杜克大学Curtarolo 等[38]发布了AFLOWlib 计算材料数据库,存储了包括无机化合物、二元合金与多元合金等超过356万种材料结构和7亿条第一性原理计算的材料性能数据,是诸多数据库中数据量最大的一个。2013 年,西北大学Wolverton 等[39]推出了开放量子材料数据库(open quantum materials database,OQMD),通过DFT计算了102万种材料的热力学和结构特性,其中以钙钛矿数据居多。以上三个数据库的数据都是从无机晶体结构数据库衍生而来,不同之处在于其所包含的虚拟材料的数量[40]。相比于国外,国内的材料计算数据库发展较晚。2016 年,北京科技大学牵头建立的“材料基因工程专用数据库”(http://www.mgedata.cn/),包含超过76 万条催化材料、特种合金及其材料热力学和动力学等数据;2020年,中国科学院物理研究所等单位创建的Atomly数据库(http://atomly.net/#matdata),包含从ICSD数据库和DFT 计算得到的18 万个无机晶体结构并计算其详细的电子结构信息以及热力学相图。这些基于计算的数据库拥有着庞大的数据量,使得数据驱动的材料研究得到迅速的发展。
然而,电化学储能材料的研发需要考虑离子输运性质、能量密度、充放电速率等特定的材料性能,上述通用数据库往往不能满足这些需求。因此,专门为电化学储能材料建立的数据库开始被研究与使用。例如,中国科学院物理研究所在2018年推出了电池材料离子输运数据库(http://eol.iphy.ac.cn/bmd/),采用键价方法计算得到了2 万多条无机晶体化合物离子迁移势垒数据,可快速筛选已知结构化合物中离子迁移势垒较低的潜在快离子导体。本课题组于2020 年发布了电化学储能材料高通量计算平台(https://matgen.nscc-gz.cn/solidElectrolyte/),集成了晶体结构几何分析(CAVD)[41]、键价和计算(BVSE)、多精度融合算法[42]和相稳定性计算等程序,并基于CAVD 和BVSE 构建了包含2.9 万条数据的离子输运特性数据库[43],能够为下游的机器学习任务提供相应的学习样本,如图2所示。为构建全面系统的电化学储能材料数据库,本团队正在引入相图计算、蒙特卡洛、相场模拟和连续介质等模块以进一步为该领域的研究提供技术支持。
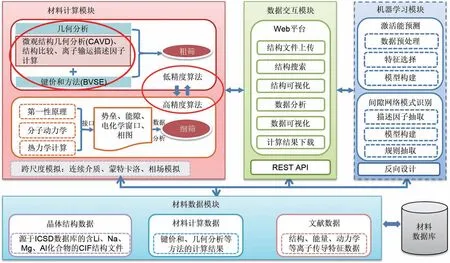
图2 电化学储能材料高通量计算平台总览Fig.2 Overview of high-throughput computing platform for electrochemical energy storage materials
综上所述,国内外各研究机构和团队建立了各种各样的通用和专用材料数据库,为数据驱动的电化学储能材料研发提供了丰富的数据资源。进一步,为支持数据驱动的电化学储能材料研发,还可以在以下三个方面对数据库建设进行完善。
第一,目前建立的电化学储能材料相关数据库收集的数据资源不够系统全面,无法满足储能电池的研发需求。一方面,在研究过程中只有小部分结果理想的数据被发表了出来,还存在大量失败实验数据并未公开,这些反例数据已经被证明能够辅助机器学习发现新材料[44]。因此,在搜集成功数据的同时,可以鼓励研究人员有针对性地将失败的实验结果保留。另一方面,现有电化学储能材料数据库中的材料数据尺度单一,储能电池的综合性能不仅与材料的本征性质相关,也与材料的微观形貌、外界环境场及器件的宏观构造等因素相互耦合[30]。因而可以建立电化学储能材料DFT计算参数库、分子动力学模拟参数库、相场模拟参数库、组分表征数据库、表界面数据库和结构表征数据库,为机器学习在电化学储能材料的应用提供多尺度数据。
第二,上述数据库主要包含了材料的结构和性能数据,通常由材料专家从中提取结构化数据或者把材料结构表示为非结构化图形数据作为机器学习模型的数据集。对于图像和文本类型的非结构化数据还无法从已有的材料数据库中获取。图像数据主要储存在材料测试机构中,通常无法公开获取。文本数据分散在各大材料科学出版物中,从海量文献中标记集成可用于机器学习的数据非常困难,且几乎没有开源具有标注信息的材料文本数据集。因此,有必要建立开源的材料图像数据库和文本数据库,推动非结构化数据驱动的电化学储能材料研发应用。
第三,对于数据的使用者来说,数据的质量决定着机器学习模型的上限。实验测量的数据质量主要受材料缺陷、污染物和实验条件以及实验设备的不确定性影响;计算模拟的数据质量主要与计算模拟方法本身的精度相关。在数据集成过程中,不同来源数据的误差相结合,使得材料数据的质量更加难以确定[45],如晶体的形成能,其计算值和实验值显著不同[46]。此外,研究人员在实验或计算过程中关注的参量具有差异性,收集材料数据时可能存在数据记录不一致的问题,造成了数据集的稀疏性。因而急需设计电化学储能材料数据质量检测方法,以提升机器学习模型的性能。
总之,通过上述方案能够优化完善材料数据资源,为材料数据和领域知识创建可持续的生态系统,从而促进数据驱动下的电化学储能新材料发现。
2 电化学储能材料数据驱动的机器学习
本节将对电化学储能材料中结构化和非结构化数据驱动的机器学习建模和应用分别进行系统地介绍,重点分析其存在的困难和挑战。
2.1 结构化电化学储能材料数据驱动的机器学习
目前机器学习在电化学储能材料领域的应用大多数是基于结构化数据驱动的,这需要针对特定的目标属性选取合适的描述符,并对其进行结构化表示,构建学习样本,进行机器学习建模和应用。具体工作流程如图3 所示。首先,可以使用实验测量、计算模拟或者直接从现有的材料数据库中收集材料原始数据,并从中提取合适的描述符,这些描述符一般包括材料结构、化学成分和材料性能等;其次,经过特征选择或者特征转换将描述符数据集转换为学习样本;然后,通过选择合适的机器学习算法并调整最优超参数,模拟条件属性与目标属性之间的映射关系;最后,研究人员可以利用这些模型来预测材料的性质或指导新材料的发现,如液态/固态电解质和电极材料的性能预测与成分优化以及电池健康状态评估。
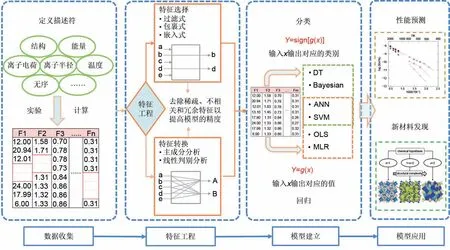
图3 结构化数据驱动的机器学习在电化学储能材料应用的工作流程[29]Fig.3 Workflow of structured data-driven machine learning in energy storage material application[29]
2.1.1 数据收集
电化学储能材料内部的微观结构与材料性能之间的关系纷繁复杂,任何一种性能都与多种因素耦合相关。从实验或者计算中收集到与目标属性相关的材料原始数据之后,还需要从中选取合适的描述符构建数据集。一般来说,相似的材料对应的描述符也要相似且数量和获取成本尽可能低[47]。然而,目前还没有普遍认可的描述符选择方法,其很大程度上依赖于研究者的领域知识。
针对特定的性能选取合适的描述符有助于建立更精确的模型,从而实现对电化学储能材料性能的精准预测。Sendek 等[48]根据原子的位置、质量、电负性和半径计算了与离子导电性相关的20 个表征晶体局域原子排列和化学环境的描述符,进而利用逻辑回归算法对锂离子电池固体电解质离子电导率的高低进行分类;赵倩等[49]基于离子传导相关因素的分析,通过整合全局及局域离子传导环境对离子传导快慢的影响,构建了一套分层编码晶体结构基描述符框架,包含组成、结构、传导通道、离子分布和特殊离子5个部分共32个描述符,并采用偏最小二乘分析(PLS)方法成功地预测了立方相Li-Argyrodites的激活能;王爱平等[50]提取了有机溶剂小分子性质、最高占据分子轨道、最低未占据分子轨道和偶极矩以及官能团的原子性质共13 个描述符,使用梯度提升决策树(GBDT)预测了溶剂与LiOH 分子的结合能,发现磷酸酯溶剂能够显著加快Li-O电池的反应动力学。这些工作都是以目标属性为导向,依靠材料专家对材料体系的认知来选取的描述符。
材料专家针对不同材料性质所选取的描述符往往不能完全通用,这导致描述符的可扩展性差。为了将无机材料原始数据转换为机器学习算法所需的学习样本,Ward 等[51]根据材料的物理和化学性质提出了一套通用的描述符计算框架,包括化学计量属性、元素属性统计、电子结构属性和离子化合物属性共145个描述符。这些描述符在电化学储能材料性能预测研究中已经得到了成功的应用[52-55]。例如,Rajendra 等[52]通过上述框架得到273 个描述符,开发了预测电极电压的机器学习模型,为钠/钾离子电池筛选了近5000 种候选电极材料;Jo 等[53]和Choi 等[54]利用上述框架和Voronoi 镶嵌方法[56]分别提取了145个化学描述符和126个结构描述符并构建机器学习模型来预测钠离子固态电解质的力学性能;Verduzco 等[55]通过选取元素属性、元素分数、化学计量属性、价轨道和实验温度共105个描述符设计了基于随机森林的主动学习方法,用于预测石榴石型固态电解质离子电导率。上述工作证明了该描述符计算框架在无机材料性能预测的适用性。
为了提高描述符的计算效率,一些研究人员开发了计算工具包对现有的描述符计算方法进行集成。如Ward 等[57]结合前期的工作基础[51]开发了基于Python 的特征生成方法库Matminer,其中包含了47 个不同的特征提取模块,能够生成数千个物理相关的描述符,大大降低了描述符计算的难度。Himanen等[58]创建了一个对原子结构进行编码的描述符库DScribe,包含库仑矩阵[59]、Ewald 和矩阵[60]、正弦矩阵[60]、多体张量表示(MBTR)[61]、原子中心对称函数(ACSF)[62]和原子位置平滑重叠(SOAP)[63]等结构描述符,并通过周期性晶体的形成能和有机分子的离子电荷预测来说明其适用性。
总的来说,上述工作的推出加速了结构化描述符的构建,为后续的机器学习模型提供了可靠的数据集。但是,目前材料样本量少且描述符的选取存在稀疏性、不相关性和冗余性导致小样本高维度问题,从而影响模型的性能。此外,尽管目前已经开发了一些集成式的描述符计算工具,但是储能材料性能影响因素的复杂性导致能够适用于任意目标属性的通用描述符提取方案还未实现。
2.1.2 特征工程
由于描述符的选择往往取决于材料专家知识,这些描述符通常存在稀疏性、不相关性和冗余性,导致模型性能较差。因此,特征工程是机器学习模型构建中的一个重要步骤,包括特征转换和特征选择。特征转换是把高维特征空间映射到低维特征空间的方法,在降低特征维度的同时特征数值也会改变。特征选择是从全部特征中选择一个特征子集,以降低样本维度,进而提高机器学习模型的预测精度和泛化性能。目前,已有学者从数据的角度利用现有的统计或机器学习方法进行纯数据驱动的特征转换或选择,试图从电化学储能材料众多描述符中挑选出材料可解释、预测精度高的描述符。
2.1.2.1 特征转换
特征转换方法主要有主成分分析[64]和线性判别分析[65]。主成分分析通过线性投影并使得所投影的维度上数据的方差最大,以降低数据集的维数、提高可解释性的同时最大限度地减少信息丢失[64]。线性判别分析是将一个高维空间中的数据投影到一个较低维的空间中,且投影后要保证各个类别的类内方差小而类间均值差别大[65]。这两种方法一般用于储能电池的系统诊断。如Banguero 等[66]将主成分分析模型应用于与电池储能系统的容量、内阻和开路电压相关的参数集处理;Wang 等[67]利用主成分分析对电动汽车动力电池一致性多参数评价;Chen 等[68]基于线性判别分析的分类模型识别锂离子电池故障。
2.1.2.2 特征选择
特征选择方法可以分为过滤式、包裹式和嵌入式三大类[69]。过滤式特征选择方法使用基于统计理论和信息论的评分标准(例如距离函数、统计相关系数和互信息等)评估相关特征的重要性并进行排序,然后在机器学习模型中使用得分高的特征子集[70],如图4(a)所示。该方法具有简单和高效的优点,然而,其特征选择过程与机器学习模型分离,忽略了所选特征子集对模型性能的影响,这通常会导致模型的预测精度较低[71]。包裹式特征选择方法首先根据预先定义的搜索策略(如穷举法,遗传算法等)生成若干初始候选特征子集,其次训练一个特定的机器学习模型来评估每个候选特征子集,保留一些候选特征子集并用于生成下一组特征子集,该过程反复进行,直到选定的特征子集满足迭代停止条件[72](模型预测精度或循环次数),如图4(b)所示。该方法能够选择出具备高精度预测性能的最优特征子集,但往往以计算时间和复杂度为代价[73]。与包裹式方法类似,嵌入式方法同样与特定的机器学习模型绑定。但不同的是,该方法通过在目标函数和建模过程中引入正则化系数或随机因素实现模型构建和特征选择的协同(例如偏最小二乘分析、LASSO和随机森林),简化了特征选择的过程,但受限于特定的机器学习模型,普适性有待提高[74]。
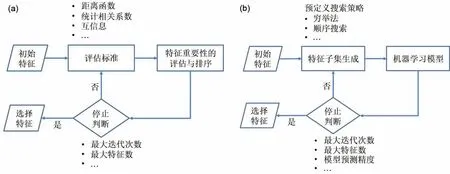
图4 特征选择方法工作流程[75]:(a)过滤式;(b)包裹式Fig.4 Workflow of feature selection method[75]:(a)filter;(b)wrapper
在电化学储能材料性能预测研究中,包裹式方法由于考虑了特征对模型性能的影响已被广泛地应用。例如,Sendek 等[48]采用穷举策略从20 个结构化描述符中选择了5个描述符,利用逻辑回归对锂离子电导率的高低进行分类;Gharagheizi 等[76]采用顺序搜索策略成功筛选出10 个关键描述符,并建立最小二乘支持向量机(LSSVM)模型预测离子液体电导率;Wu 等[77]利用顺序搜索方法从111 个描述符中选择了23 个关键描述符,采用高斯核岭回归模型预测FCC 溶质扩散势垒。嵌入式方法在选择特征的同时可以根据特征的重要性进行排序,使得专家可以更有针对性地进行材料设计,对于电化学储能材料的研究有着重要意义。例如,Shandiz等[78]为339 条硅酸盐阴极材料样本构建了9 个描述符,利用极大随机化树(ERT)预测其晶系结构,发现晶胞体积是最重要的特征。赵倩等[49,79]通过分层编码晶体结构描述符为50 条立方相Li-Argyrodites样本构建了32 个描述符,并借助偏最小二乘分析(PLS)方法推断各描述符与激活能之间的因果关系。
另外,过滤式和包裹式方法组合也是一种有效的特征选择方法,这种方法可以从数据的不同角度对特征进行处理[80]。例如Hsu 等[81]先通过计算效率高的过滤器从原始数据集中选择候选描述符,然后通过更准确的包裹器进一步优化得到训练样本。在电化学储能材料研究领域,刘悦等[75]首次提出了一种融合加权评分领域专家知识的多层级特征选择方法,其方法框架如图5所示。该方法将过滤式和包裹式方法相结合自动去除稀疏、不相关和冗余特征,在特征选择过程中引入领域专家知识,消除了关键特征被删除的风险,并在四个电池材料数据集上进行了实验,显示出比其他方法更好的预测性能。

图5 融合加权评分领域专家知识的多层级特征选择方法框架[75]Fig.5 Multi-level feature selection method framework combining weighted scoring domain expert knowledge[75]
总之,许多结构化数据不仅维数高且样本量小,导致机器学习模型的过拟合,降低了模型的泛化能力。这也是电化学储能材料科学中需要特征工程的重要原因。然而,由于特征选择方法复杂多样,且涉及的超参数和策略也需要手动设置和调整。例如过滤式方法需要设置所选特征的数量和过滤阈值;包裹式方法需要指定子集搜索策略以生成候选特征子集;嵌入式方法需要优化机器学习算法的超参数以获得更好的性能。这将导致没有相关经验的材料专家不易使用这些方法。另外,特征工程仅仅通过特征空间的分布来选择描述符,这可能使得一些关键描述符重要度被弱化,导致学习结果与领域知识不一致。
2.1.3 机器学习建模与应用
目前,机器学习在储能电池领域得到了广泛的应用,其优越性在时间效率和预测精度上都得到了证明。其中,各种算法具有不同的特点和适应范围,选择合适的机器学习算法是构建机器学习模型的关键步骤,这极大地影响了其预测的准确性和泛化能力[82]。当前常用于储能电池研发的机器学习方法如表2所示。下面介绍这些方法在储能电池应用中的最新进展。

表2 常用于储能电池研发的机器学习模型对比Table 2 Comparison of machine learning models commonly used in energy storage battery research and development
2.1.3.1 液态电解质研究中的应用
液态电解质是电池的重要组成部分,它在正负极之间传输离子的同时也起着阻碍电子传导的作用,对电池的性能至关重要[96]。机器学习已经被成功用于液态电解质化学稳定性、离子与溶剂的配位能预测以及溶剂成分优化。化学成分之间的稳定性和兼容性是在配置电解液时需要考虑的基本参数,Lee 等[93]将机器学习方法与传统反应指数相结合开发了神经网络回归模型并准确预测了93 种电解质溶剂和氧化还原介质之间化学稳定性。离子与溶剂的配位能是影响离子输运的重要因素之一[97],Ishikawa等[89]计算了70种溶剂分别与5种碱族元素的配位能,选择了13 个离子和溶剂相关描述符并采用高斯过程回归预测元素配位能。电解质添加剂及其成分的优化组合是实现高压电池长循环性能的有效方法,Duong等[94]选择电解质添加剂比例、负极和正极的容量比及循环次数作为输入参数,使用人工神经网络模型预测电池容量并成功地找到性能优异的电解质成分。
2.1.3.2 固态电解质研究中的应用
近年来,固态电解质因比液态电解质具有更好的安全性、更高的能量密度和更长的寿命备受关注[1]。利用机器学习对其离子电导率、化学组成和带隙进行预测是一个研究热点。离子电导率是衡量一种材料是否可以用作固体电解质的重要指标之一,Xu 等[83]收集127 种实验合成的NASICON 和LISICON材料并利用包裹式特征选择方法选取7个简单描述符,然后通过逻辑回归模型预测离子电导率。提高材料离子电导率的常见策略是掺杂添加剂或混合异质材料,Homma 等[90]通过实验制备了15 个多相三元Li3PO4-Li3BO3-Li2SO4混合物样本,使用高斯过程回归的贝叶斯优化成功找到离子电导率性能优异的三元相化学组成比例。此外,带隙也是影响固体电解质性能的关键因素,Wang 等[92]从Materials Project 数据库中收集了286 个具有计算带隙的石榴石结构并选取28 个描述符来训练XGBoost 模型,最后筛选出12 个潜在的石榴石型固态电解质材料。
2.1.3.3 电极材料研究中的应用
电极材料的平均电压、体积变化、界面反应能、初始放电容量、库仑效率和电极制造参数对电池综合性能有着重要的影响,通过实验和计算来获得这些性质总是困难和昂贵的,因此有必要通过机器学习对其进行预测。电极材料的平均电压和充放电时的体积变化分别影响着电池的能量密度和安全性能,Moses 等[95]从Materials Project 数据库收集了4860个材料,通过电极的化学计量以及Matminer工具包生成了306个描述符,使用神经网络模型预测电极材料充放电时的平均电压和体积变化。为了探寻锂金属负极的枝晶生长和高反应性导致电池循环效率低和安全性差的原因,刘波等[84]计算了100 种LLZOM 化合物的界面反应能,将掺杂元素的15 个相关特性视为描述符,通过支持向量集和核岭回归模型对界面稳定性和反应能进行准确预测。富锂层状氧化物正极材料在充放电过程中由于不可逆相变导致其结构稳定性降低、容量衰减和电压下降,Kireeva 等[85]收集了99 种富锂层状氧化物正极材料,选择化学成分、原子性质、合成方法和实验条件作为描述符,使用支持向量机模型成功预测了富锂层状氧化物的初始放电容量和库仑效率。此外,由于电极的制造过程、电极浆料特性和涂层参数强烈影响电池的性能和寿命,Duquesnoy等[88]制备了144 个涂层电极,通过主成分分析、K均值聚类和高斯朴素贝叶斯分类器方法相结合,从而预测了与特定制造参数相关的电极异质性。
2.1.3.4 电池健康状态评估中的应用
电池健康状态的评估对于电池系统的平稳可靠运行至关重要。而电池老化是一个复杂的过程,涉及阳极、阴极和电解质/电极界面的许多电化学反应。另外,温度和负载等操作条件也会影响电池老化过程[98]。目前机器学习技术已被成功应用于预测电池的容量和健康状态,以确保设备可靠运行和及时维护。Nagulapati 等[86]使用18650 个电池充放电数据,利用高斯过程回归和支持向量机模型将放电循环过程中的电压、电流和温度数据相关联预测电池容量,并通过组合多电池数据集的方法提高了模型的预测精度。与常用的电流-电压数据相比,电化学阻抗谱通过测量电流对电压扰动的响应来获得宽频率范围内的阻抗,包含有关材料特性、界面现象和电化学反应的丰富信息。Zhang等[91]收集了2万多个商业锂离子电池的电化学阻抗谱,使用高斯过程回归模型将整个阻抗谱作为输入,准确预测了不同温度下电池健康状态和剩余使用寿命。大幅度延长电池使用寿命的关键挑战是减少所需实验的数量和持续时间,Attia等[99]通过弹性网络早期模型结合贝叶斯优化算法,从前100个循环的电化学测量数据预测了最终循环寿命并有效地探测充电协议的参数空间。
综上所述,通过提取材料的结构特征、元素属性和实验环境等结构化描述符建立机器学习模型,能够指导研究人员设计和优化液态/固态电解质和电极材料以及评估电池的健康状态。然而,由于其内部复杂的电化学行为,电化学储能材料的微观结构和材料性能之间常常存在复杂的非线性关系,导致线性模型性能较差,而神经网络等非线性模型的复杂性高可解释性差,且一般需要大量数据进行训练。因此,还需要研究人员对结构化数据驱动的电化学储能材料研发进行进一步的探索。
2.2 非结构化电化学储能材料数据驱动的机器学习
随着对电化学储能材料的深入研究,能够通过材料结构、表征技术和科学文献中得到大量的图形、图像和文本等非结构数据。这些数据含有非常丰富的材料信息,但传统的机器学习方法难以对其处理。作为机器学习的重要组成部分,深度学习能够从非常原始的非结构化图形和图像数据中提取信息,实现自动模型参数估计(即“端到端”学习),从而避免繁琐但重要的描述符设计[100-101]。此外,通过文本挖掘和自然语言处理技术能够从非结构化的文本中提取材料的数据和知识,为进一步数据挖掘和分析提供数据集。
2.2.1 基于图形的材料性能预测
近年来,图深度学习(graph deep learning)因能够对任意大小和形状的图形进行“端到端”学习,无需研究人员构建繁琐而重要的描述符受到了材料领域越来越多的关注。
起初,图深度学习被应用在分子体系的性能预测中[102-103],随后被用于周期性晶体体系[104-108]。工作流程如图6 所示,首先需要获取材料结构数据集,一般来源于ICSD数据库或CSD数据库;然后对材料结构进行图形表示,这个过程需要选择合适的材料信息进行编码,如原子轨道相互作用、原子属性、键属性、全局状态和阴离子配位多面体基序等;最后通过图深度学习模型预测材料性能。这里总结了不同材料信息嵌入下图深度学习在材料性能预测中的应用。

图6 图深度学习在分子/晶体结构应用的工作流程Fig.6 Workflow of application of graph deep learning in crystal and molecular structure
2.2.1.1 原子信息嵌入
最近大多数材料图深度学习研究是基于原子级别的图形数据作为深度学习模型的输入[104,107-108]。在谢天等[104]提出的晶体图卷积神经网络(CGCNN)中,每个晶体由一个晶体图形表示,并且满足原子索引置换不变性和晶胞选择不变性,该模型准确地预测了晶体结构的形成能、带隙、费米能和弹性特性等性能,最后通过钙钛矿材料说明了模型的可解释性。Ahmad 等[109]应用CGCNN 模型筛选能够抑制锂金属阳极枝晶形成的无机固体电解质。Zhou等[110]基 于CGCNN 模 型 从Materials Project 和AFLOW 材料数据库中筛选了13 万多种无机材料,成功预测了80 种可用于锌离子电池的高压正极材料。作为CGCNN 的改进模型,Park 等[108]提出了iCGCNN 模型,该模型的晶体图包括Voronoi 镶嵌晶体结构的信息、相邻组成原子的显式三体关联以及化学键的表示,在预测热力学稳定性方面,iCGCNN的精度相较于CGCNN提高了20%。
2.2.1.2 原子和全局状态信息嵌入
由于之前绝大多数模型是单独基于分子或晶体数据集开发的,且缺乏对温度和压力等全局状态的描述,致使模型缺少必要信息影响其预测性能。因此,Chen 等[107]提出了一个基于图形的深度学习框架(MEGNet)并将其应用于分子和晶体结构。该模型通过将原子属性、键属性和全局状态属性嵌入图神经网络模块中,然后通过信息传递过程反复更新,最终利用多层感知器预测材料性能。MEGNet在预测晶体的形成能、带隙和弹性模量方面显著优于现有的机器学习模型。在此基础上,该团队还开发了一个能够处理多保真度数据和无序材料的图神经网络模型[111]。该模型将数据保真度级别编码为整数并传递给可训练的保真度嵌入矩阵,并通过元素嵌入的线性组合表示无序位点。实验结果表明该模型对于实验带隙预测的平均绝对误差降低了22%~45%,但潜在限制是它依赖于大型低保真数据集来学习有效的结构表示,导致只能对少数目标属性进行高精度预测。
2.2.1.3 多尺度材料信息嵌入
与弹性模量等力学性能相比,预测晶体材料的电子结构性质需要更详细的材料信息,因此仅嵌入原子信息的图深度学习模型可能性能不佳。Banjade等[106]提出了一种Atom-Motif双图网络模型(AMDNet)以增强对电子结构相关材料性能的预测。该模型利用阴离子配位多面体构建结构基序图,及基于原子的图形一起输入神经网络模型。与已有的模型相比,AMDNet 预测金属氧化物带隙等性能更加准确。此外,原子轨道也是一种与电子结构高度相关的材料特征。Karamad 等[105]提出了一种轨道图卷积神经网络(OGCNN),以考虑晶体材料的原子轨道信息。该模型将原子轨道之间的键合信息编码为轨道场矩阵(OFM)表示,然后将改进的节点和边特征传递给CGCNN框架以进行性能预测,最后在形成能和带隙预测方面都具有比CGCNN 更好的性能。
综上所述,本文对图深度学习的图形表示方法和模型框架进行了介绍,这些框架能够对材料的性能进行高效准确地预测,加快材料的研发速度。然而其中还存在一个基本问题:通过简单地设置截断距离可能导致原子间距离的微小变化使邻居原子数量的突然变化,晶体结构中原子的连接性难以判断。因此,图形表示更适合于具有共价键的分子材料,而具有离子键和金属键的晶体材料需要特别注意识别节点的连接性[112]。
2.2.2 材料表征图像分析
材料内部的显微组织结构决定着材料的性能,通过现代材料分析技术对其进行表征,可得到图像类型的非结构化数据。这些数据通常需要依赖材料专家对其进行分析,从中提取出显微化学成分、晶体结构和微观形貌等材料信息。但是仅仅依赖材料专家自身经验分析容易遗漏其中的隐藏信息,且耗时费力。计算机视觉领域的深度学习方法可以自动提取图像中的特征,与材料图像数据分析的强烈需求相吻合,有助于提高材料表征的速度和准确性。这里主要针对不同的材料表征技术,介绍深度学习在电化学储能材料图像数据分析的研究进展。
2.2.2.1 X射线断层扫描图像分割
X射线断层扫描是一种强有力的表征方法,可以对材料的微观结构和化学成分进行动态无损成像,提供电池运行和退化的定量或定性分析[113]。量化锂电极中微结构的形态转变需要严格和一致的分割程序,Dixit等[114]实现了一个基于ResNet-34的深度卷积神经网络对锂金属X射线断层扫描低对比度图像中的锂金属和孔隙进行分割,以定量跟踪锂金属电极和固态电解质固固界面的形态变化。与传统的二值化过程相比,机器学习识别锂金属孔隙特征的保真度和准确性明显提高。复合电极的微观结构决定了电极颗粒在充放电过程中的行为,颗粒与碳/黏合剂分离的程度与容量损失相关。为了对严重破碎的颗粒进行识别,Jiang 等[115]使用高分辨率硬X射线纳米断层扫描对复合正极材料可视化,开发了一个掩模区域卷积神经网络模型并自动识别和分割了650多个正极颗粒,消除了使用传统图像技术报告中表征结果存在的偏差。
2.2.2.2 拉曼高光谱成像特征提取
拉曼高光谱成像具有同时对多种化学特征进行成像的能力。同步识别锂离子电池电极中多个光谱特征有助于将分析技术用于在线质量控制和产品开发。Baliyan 等[116]提出了一个神经网络分析框架来自动从锂离子电池电极拉曼高光谱数据集中识别光谱特征并分配类别标签,从而计算容量保留系数来定量评估锂离子电池的容量退化。该方法有效地避免了宇宙噪声带来的错误定量分析,且实现了对高光谱分析整个生命周期的自动化处理。
2.2.2.3 电子背散射衍射图像晶界增强
电子背散射衍射通过分析晶粒两侧像素之间的取向来检测多晶样品中的晶界,可以在晶粒尺度上改善正极材料的评估和量化,这对理解锂离子电池的锂传输、速率限制和降解机制至关重要[116]。Furat 等[117]使用电子背散射衍射技术对正极材料颗粒进行成像,通过卷积神经网络对标记的图像进行训练并应用于整个图像数据,从而产生具有增强晶界的新图像。该方法避免了常规图像处理方法繁琐的处理步骤和参数校正过程,实现了晶粒结构的有效形态表征。
总之,深度学习技术能够从复杂的电化学储能材料图像数据中识别特定的特征,从而有效应用于X射线断层扫描图像分割、拉曼高光谱成像特征提取和电子背散射衍射图像晶界增强。然而,深度学习模型强烈依赖于大量标记的图像数据,以及非专业研究者对深度学习模型使用的复杂性等问题还阻碍着其在材料图像领域的应用。此外,上述例子仅仅是对电化学储能材料图像本身进行了建模应用,通过深度学习技术还可以进一步地探索化学成分-介观尺度显微组织结构-材料性能之间的构效关系,加速材料性能预测[118]。
2.2.3 材料文本挖掘
文本挖掘是指从文本语料库中提取有价值信息和知识的方法。近年来,材料科学的文本挖掘主要依靠自然语言处理技术和机器学习方法,从数量庞大且不断增长的科学出版物中快速获取非结构化科学知识,进而指导材料相关领域的研究。文本挖掘的工作流程可以概括为文本收集与解析、文本预处理、文本分析、信息提取、数据挖掘,如图7 所示[119]。随着文本挖掘技术的逐渐成熟,已有学者将其应用到电化学储能材料领域,从而追踪材料研究动态、指导材料合成和建立材料数据库等。
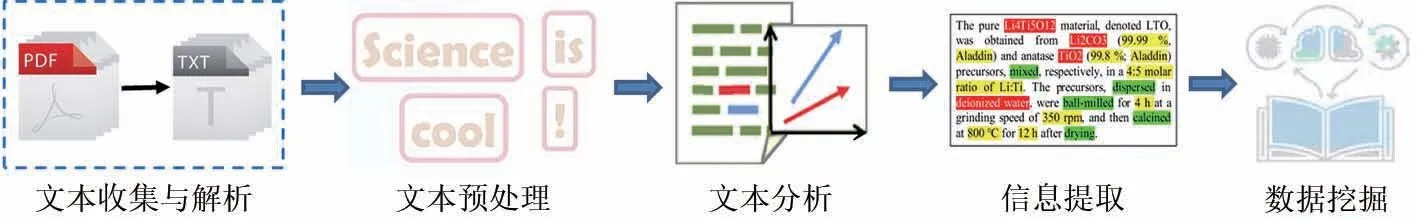
图7 文本挖掘的工作流程[119]Fig.7 Workflow of text mining[119]
2.2.3.1 追踪研究动态
文本挖掘可帮助读者找到某个领域的突破性论文并跟踪最新技术的进展。Torayev 等[120]使用基于机器学习的文本挖掘技术从1800 多篇文献中识别Li-O2电池研究领域的全球趋势。结果显示,该领域的电解质研究已从碳酸盐转向了甘醇二甲醚和二甲基亚砜,且大部分文献都关注电池的循环稳定性、容量和倍率性能。El-Bousiydy 等[121]使用基于关键字搜索文本挖掘算法,分析了1.3 万份锂和钠离子电池科学文献中研究人员的习惯,发现大多文献缺乏对某些关键特征的系统报告,例如厚度、孔隙率、电解质体积、表面积和质量载荷。通过文本挖掘技术构建材料知识图谱,能够从海量材料科学文献中进行信息抽取,建立实体之间的对应关系,从而自动化地提供材料科学领域信息。Nie 等[122]收集了超过290 万篇材料领域的文章及其作者信息,结合机器学习和依赖匹配算法对材料知识图谱中的主体进行高精度消歧,并使用剪枝策略实现高效信息匹配和搜索,从而构建了材料知识图谱(MatKG)框架。利用该框架对LiFePO4进行自动化分析,关联相关学者及其研究信息,建立了用于锂离子电池的LiFePO4材料发展里程碑图。
2.2.3.2 指导材料合成
优化电解质低温处理协议能够最大程度地减少电池界面的不兼容性[123]。Mahbub等[124]使用基于规则和机器学习方法自动提取硫化物和氧化物的锂固态电解质文本中实验合成部分,然后通过神经网络模型对每个段落中的单词进行标记和分类,以预测句子中每个单词的重要合成关键词(例如材料名称、操作名称、数量、条件等),将这些分类的标记组合成一个数据库对象并对其进一步数据挖掘以提取合成趋势。该团队从中识别出高电位氧化物基锂石榴石电解质的低温合成方法,降低了固态电解质组装到电池过程中的界面复杂性。
2.2.3.3 建立材料数据库
化 学 感 知 自 然 语 言 处 理 工 具 包ChemDataExtractor[125]是化学信息提取和文本处理的常用工具,在文本处理、标记化和词性标注方面灵活而准确,能够用于识别化学物质实体、相关属性及其相互依赖关系。大型电池材料数据库对于数据驱动的新材料发现至关重要,Huang 等[126]使用ChemDataExtractor,通过文章检索、数据提取、数据清理、数据后处理和评估过程,从22 万余篇电池研究论文中自动提取数据,然后创建了一个大型电池材料同源属性数据库,包括1.7 万种化合物和对应的21万多条电池材料属性(容量、电压、电导率、库仑效率和能量)。
综上所述,目前只有少数基于文本挖掘的工作专注于电化学储能材料领域,其限制主要有以下三点:一是材料文本标注数据稀缺性,大多数现有的标注数据集都是以特定的材料领域而创建的,难以直接应用于其他材料体系;二是材料命名方法差异性,材料文本中存在各种专业术语,缺乏标准的命名方法容易导致歧义的产生;三是材料文本的复杂性,材料科学文本的专业性强可读性差,使得文本处理异常困难。即使如此,随着大型材料文本数据库的建立和自然语言处理技术的发展,相信文本挖掘技术会对电化学储能材料的发展起到重要的作用。
3 电化学储能材料机器学习面临的挑战与对策
如前所述,结构化和非结构化数据驱动的机器学习模型已经在电化学储能材料领域得到了广泛应用,但仍存在一些问题制约着机器学习的进一步发展。本节对这些问题进行了系统性分析,并将其归结为机器学习在电化学储能材料领域应用面临的三大矛盾,包括高维度和小样本数据的矛盾、模型复杂性和易用性的矛盾以及学习结果与领域知识的矛盾。调和这些矛盾以提升机器学习模型在电化学储能材料领域应用的准确性、易用性和可解释性,将有助于进一步加速电化学储能材料的研发与设计。
3.1 高维度与小样本数据的矛盾与协调
电化学储能材料数据通常是多源(如实验数据、计算数据、生产数据和文献数据)且异构的(如结构化和非结构化数据),不同来源数据的外部一致性很难得到保证,导致最终用于机器学习建模的数据集往往是小样本的。尤其是文献数据的标记难,小样本问题更显突出。此外,电化学储能材料性能受多种物理/化学因素影响,研究人员总是定义大量描述符来表示复杂的材料性能驱动机制,这又导致机器学习建模使用的数据集通常是高维度的。较小的数据量和较高的维度容易使得机器学习模型过度拟合现有数据,从而影响机器学习模型的泛化性能和可解释性,故电化学储能材料领域机器学习面临着高维度和小样本数据的矛盾。
正如第2节中指出的,从数据的源头抓起,注重反例数据的收集、多尺度数据的收集,积累更加丰富多样的结构化和非结构化数据。同时,提高各种数据的集成与共享,克服其质量参差不齐、数据标注不足、缺乏有效融合以及大型标准数据集建设不足等问题,是调和上述矛盾的可行和必经途径。从技术上来说,目前普遍的做法是通过特征工程或选择适用于小样本高维度数据的学习器来解决上述问题。其中,如3.1.2 节所述,特征工程方法旨在通过降低特征维度或构建“更好”的描述符来削弱高维度数据集对机器学习模型性能的负面影响。支持向量机是通过核函数将低维数据投影到高维空间中进行划分,在形式上更容易处理高维特征的数据集[127]。例如,Fujimura 等[87]利用支持向量机对LISICON 型固态电解质材料的离子电导率进行预测,平均绝对误差为0.373 S/cm。然而,在处理一些复杂的小样本高维度数据问题时,这些方法的效果并不理想。因此,研究人员提出数据增强、主动学习和迁移学习等方法解决电化学储能材料数据的小样本问题来调和这一矛盾。数据增强技术基于现有数据,通过物理增强、统计学和无监督生成模型等方法生成更多新数据[128]。例如,Naaz 等[129]设计了一种基于生成对抗网络的数据增强方法用于预测锂离子电池的充电状态和健康状态;Hsu等[130]利用生成对抗网络框架来学习和生成固体氧化物燃料电池电极的微观结构。主动学习利用预构建的机器学习预测模型迭代地对候选化学空间进行自适应采样,从而为代价高昂的模拟计算或实验验证提供最有价值的候选样本,以加速新型高性能材料的筛选[131]。例如,Verduzco 等[55]利用主动学习方法指导高离子电导率的石榴石的合成,从而减少了30%的实验次数。迁移学习旨在通过迁移包含在相关领域中的知识来提高目标学习器在目标领域上的性能,以减少目标学习器对目标数据集大数据量的依赖[132]。例如,Wang 等[133]基于CGCNN 的迁移学习模型在低精度PBE 数据集预训练,到高精度HSE06 数据集进行参数微调预测晶体结构的高精度带隙,从而克服了小数据集导致的精度低和过拟合的问题。
3.2 模型复杂性与易用性的矛盾与统一
机器学习的最初目标是从数据中提取可解释的知识,并在追求算法准确性的同时强调其可解释性[127]。以线性回归、偏最小二乘法等多元线性模型为主的机器学习算法可以构建多个因素与目标属性之间的线性关系,模型简单、易于实现且学习结果容易理解。但是,电化学储能材料内部复杂的电化学行为导致线性模型的预测精度通常较低,而神经网络(NN)和支持向量机(SVM)等模型由于能够建立影响因素与目标性能间的复杂非线性关系,在电化学储能材料研究中得到了更广泛的应用。然而,这些非线性模型大都是“黑箱”模型,其内部原理复杂、结果难以解释,且通常需要大量繁琐的调参工作才能获得最优性能。因此,机器学习在电化学储能材料领域的应用中存在着模型复杂性和易用性的矛盾。
降低材料专家使用模型的复杂性和提高复杂模型的可理解性是提高模型易用性的有效途径。自动机器学习(auto machine learning,AutoML)是在有限的计算资源内全部或部分配置适用于机器学习方法的参数,主要通过随机搜索、进化优化、贝叶斯优化、元学习等方法减少模型的选择、优化以及实现过程中的人工参与,从而自动构建最佳机器学习模型[134-135]。例如,Dunn 等[136]提出自动机器学习模型Automatminer 以预测无机固体材料的性能。该模型利用Matminer[57]生成特定材料的描述符,自动地执行数据预处理和特征工程,并通过广泛的内部数据测试来确定最佳机器学习模型。而机器学习的可解释性是机器学习模型以人类可理解术语向人类提供解释的能力。在计算机领域,机器学习的可解释性可使得机器学习模型的结构和预测结果两方面都易于理解,从而提高机器学习模型的易用性。例如:规则提取方法采用“如果输入特征x则被归类为y类”的解释方式,将机器学习中隐含的知识以一种易于理解的方式表达,以提高机器学习方法的可解释性[137]。目前,可解释性方法还没有在电化学储能材料领域得到应用。未来可以引入对模型决策过程解释的内部解释和对模型决策结果解释的外部解释来共同提高机器学习应用方法的可解释性。内部解释一般把已训练好的机器学习/深度学习等模型的内部决策结构(如网络结构、参数权重、特征向量等)映射成易于理解的If-Then-Else规则或内部决策过程的权重可视化展示[138]。外部解释可将不同的特征组合输入到已训练好的模型,来找到输入特征与模型决策结果之间的关系,以发现模型的决策规则来提高模型的可解释性;也可将更复杂的模型转换为易于理解的模型,再从中抽取规则,使得抽取的规则对模型有重现能力[139]。此外,根据提取的规则构建概念嵌入表达,再将规则表达嵌入其他模型中,也有望在保证预测精度的前提下,提高模型的可解释性。
3.3 模型学习结果与领域专家知识的矛盾与融合
目前,广泛应用于电化学储能材料发现和性能预测的机器学习模型大都是纯数据驱动的,严重依赖于样本数据进行学习,对材料领域知识的重视度不够,导致在实际应用中仍然会出现机器学习结果与领域专家知识相矛盾的现象。针对该问题,一方面,可以通过描述符定义和选择过程[75]将材料专家的领域知识融入到问题定义中,从而指导模型学习领域知识。例如,Li 等[140]提出了“中心-环境”(center-environment,CE)特征构建模型,通过将基本属性集合映射到由组分和结构信息组成的基集中来构建特征,用于预测尖晶石氧化物的形成能、晶格参数和带隙;Weng等[141]利用符号回归得到了描述符μ/t(μ为八面体因子,t为容忍因子),并在该描述符的指导下成功地合成了五种新的氧化物钙钛矿;Gong 等[142]通过机器学习和理论模型相结合来预测二维金属材料上的锂吸附能,提高了模型的泛化能力。另一方面,在机器学习模型构建过程中嵌入领域知识是一个有效的解决方案,典型的算法有贝叶斯网络和模糊学习。其中,贝叶斯网络通过在训练过程中结合先验知识来确定网络拓扑结构[143],而模糊学习则使用隶属函数来整合专家经验[144]。例如,Ren等[145]通过施加基于物理场的约束来创建参数化过程模型,从而将过程优化变量与所得材料的体积和界面特性耦合起来;再添加额外的推理层将过程变量和材料属性之间的联系扩展到器件性能,并利用神经网络代理模型预测电流电压曲线;最后,通过贝叶斯网络推断结果优化太阳能电池工艺参数。此外,可通过机器学习结果建立知识库与电池材料专家先验知识共同指导材料开发。例如,Martin等[146]提出一种将领域专家与机器学习相结合构建知识库的方法来实现两者的相互补充,从而提高专家系统的推理能力。
目前,绝大部分的机器学习方法是纯数据驱动的,机器学习的全过程仅围绕着提升模型精度这一单一目标展开,往往忽略了领域知识的重要性,这是导致上述三大矛盾的主要原因之一。基于此,为充分发挥材料领域知识在机器学习建模中的作用,本团队提出的融合加权评分领域专家知识的多层级特征选择方法[75]和分而治之的自适应机器学习建模方法[147],已经初步证明了领域知识嵌入在改善机器学习模型预测精度和可解释性方面的有效性。进一步地,将领域知识符号化表示为机器学习模型的前处理条件、建模约束或目标函数、后解释规则等并嵌入到机器学习全生命周期过程中,同时结合知识、数据、算法和算力四大要素,构建具有一定可解释性的领域知识嵌入的机器学习新模型,将有望系统性地解决上述三大矛盾。
4 结 语
数据驱动材料科学的最新研究表明,机器学习技术的应用可以极大地促进电化学储能材料的设计和发现。本文首先介绍了可用于电化学储能材料研究的数据资源,并对电化学储能材料专用数据库发展方向提出建议,如收集实验反例数据和材料多尺度数据、共享图像和文本等非结构化数据、设计数据质量检测方案;随后,详细阐述了结构化数据驱动下的机器学习工作流程及其在电化学储能材料领域的最新应用,以及基于图形、图像、文本的非结构化数据驱动下的机器学习在电化学储能材料领域的研究进展;最后,总结了机器学习在电化学储能材料领域应用所面临的三大矛盾和相关的解决策略,并提出进一步构建面向机器学习全流程的“领域知识嵌入的机器学习方法”,将有望系统地调和上述三大矛盾。本文对机器学习在电化学储能领域应用的总结和未来发展策略的提出,将为实现高性能电化学储能材料的精准、高效研发指明方向。
猜你喜欢
矿山安全信息(2022年9期)2022-11-24
环球时报(2022-07-13)2022-07-13
电子乐园·下旬刊(2022年5期)2022-05-13
环球时报(2022-03-14)2022-03-14
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
股市动态分析(2021年7期)2021-04-20
电影(2018年8期)2018-09-21
广东教育·高中(2018年12期)2018-02-13
分析化学(2017年12期)2017-12-25
试题与研究·高考理综化学(2016年4期)2017-03-28
