
婴幼儿配方乳粉中氯丙醇酯含量测定方法的研究
2022-03-10 12:09:48张红霞卢克刚梁秀清陈倩倩王艳丽刘艳明祝建华
中国酿造 2022年2期
张红霞,卢克刚,梁秀清,陈倩倩,王艳丽,刘艳明*,胡 梅,祝建华
(1.山东省食品药品检验研究院 山东省特殊医学用途配方食品质量控制工程技术研究中心,山东 济南 250101;2.山东职业学院,山东 济南 250104)
氯丙醇酯(chloropropanol esters)是近年来备受关注的一类食品污染物,按氯丙醇种类不同可分为单氯取代的3-氯-1,2-丙二醇酯(3-monochloropropane-1,2-diol ester,3-MCPDE)和2-氯-1,3-丙二醇酯(2-monochloropropane-1,3-diol ester,2-MCPDE)以及双氯取代的1,3-二氯-2-丙醇酯(1,3-DCPE)和2,3-二氯-1-丙醇酯(2,3-DCPE)[1-2]。虽然氯丙醇酯自身的毒性尚无定论,但在人体脂肪酶作用下可水解为氯丙醇,氯丙醇具有致癌性、致突变性、肾脏毒性和生殖毒性[3-4]。研究发现,氯丙醇酯广泛存在于玉米油、芝麻油、菜籽油和棕榈油等[5-8]精炼植物油以及烘烤食品和油炸食品等[9-11]热加工食品中,其中检出率较高的为单氯取代的3-MCPDE与2-MCPDE,而双氯取代的氯丙醇酯少有检出[12-14]。婴幼儿配方乳粉在生产工艺中为了匹配母乳中脂肪的含量和组成,通常添加玉米油、大豆油、棕榈油等精炼植物油作为外源性脂肪补充,这就导致了配方乳粉中可能存在氯丙醇酯暴露风险,为了保障婴幼儿饮食健康安全,婴幼儿配方乳粉中氯丙醇酯(尤其是3-MCPDE与2-MCPDE)的分析研究备受国内外关注[15-16]。
目前氯丙醇酯含量的测定多采用间接测定法,即通过水解将氯丙醇酯转化为氯丙醇,以氯丙醇的含量表征氯丙醇酯的含量[17-20]。婴幼儿配方乳粉的样品处理方法通常有两种:一是从乳粉样品中直接提取氯丙醇酯(直接提取法)[21-23];二是先提取乳粉样品中脂肪再测定脂肪中的氯丙醇酯(提取脂肪法)[16,19,24-25]。提取脂肪法前处理过程繁琐,溶剂使用量大,不易批量化检验。
该研究以婴幼儿配方乳粉为研究对象,在GB5009.191—2016《食品安全国家标准食品中氯丙醇及其脂肪酸酯含量的测定》第三法的基础上优化改进,比较了直接提取法和提取脂肪法两种提取方法、苯硼酸衍生和七氟丁酰基咪唑衍生两种衍生方法的差异,以期建立简便、快速、结果准确、灵敏度高的检测方法,实现样品的批量化处理,提高实验室检验效率,为婴幼儿配方乳粉中氯丙醇酯污染的数据分析和风险监测提供技术支持。
1 材料与方法
1.1 材料与试剂
婴幼儿配方乳粉样品:市售;3-氯-1,2-丙二醇棕榈酸二酯、2-氯-1,3-丙二醇硬脂酸二酯、3-氯-1,2-丙二醇、2-氯-1,3-丙二醇、D5-3-氯-1,2-丙二醇、D5-2-氯-1,3-丙二醇(纯度均≥98%):北京曼哈格公司;D5-3-氯-1,2-丙二醇棕榈酸二酯、D5-2-氯-1,3-丙二醇硬脂酸二酯(纯度均≥98%):加拿大TRC公司;苯硼酸、甲醇钠、七氟丁酰基咪唑(纯度均为97%):上海阿拉丁公司;正己烷、甲醇、乙酸乙酯、叔丁基甲基醚、异辛烷、乙腈(均为色谱纯):美国Fisher公司;乙醚、石油醚、氨水、乙醇、溴化钠、浓硫酸、氯化钠、无水硫酸钠(均为分析纯):国药集团化学试剂有限公司。
1.2 仪器与设备
7890B-5977A气相色谱-质谱联用仪:美国Aglient公司;R300旋转蒸发仪:瑞士Buchi公司;MS3涡旋混合振荡器:德国IKA公司;N-EVAP-45型氮吹仪:美国Organomation公司;3K15型高速离心机:德国Sigma公司;Milli-Q IQ7000超纯水机:美国Millipore公司。
1.3 方法
1.3.1 溶液配制
分别称取适量的氯丙醇酯、氯丙醇及内标于10 mL的容量瓶中,用乙酸乙酯溶解并定容,制备质量浓度为1 000 mg/L(以氯丙醇计)的标准品单标储备液。用乙酸乙酯稀释成质量浓度均为100 mg/L(以氯丙醇计)的标准品单标中间液。分别吸取适量的标准品单标中间液,用乙酸乙酯稀释成质量浓度均为2 mg/L(以氯丙醇计)的酯混合标准工作液、酯混合内标工作液、醇混合标准工作液和醇混合内标工作液。所有标准溶液均在-18 ℃条件下保存。
甲醇钠-甲醇溶液(25g/L):称取甲醇钠2.5g,加入100mL甲醇,搅拌使其充分溶解。酸化溴化钠溶液(600 g/L):称取60 g溴化钠溶解于100 mL水中,加入3.5 mL 25%的硫酸。乙醚-乙酸乙酯溶液(6∶4,V/V):量取60 mL乙醚,加入40 mL乙酸乙酯,混匀。苯硼酸溶液:称取200 mg苯硼酸,加入10 mL乙醚,涡旋,制成苯硼酸饱和的乙醚溶液,取上清液使用。
1.3.2 样品前处理
(1)提取
直接提取法:称取0.5 g乳粉样品(精确到0.1 mg)于15 mL聚丙烯离心管中,加入50 μL质量浓度为2 mg/L(以氯丙醇计)的酯混合内标工作液,加入5 mL温水,涡旋混匀,加入约1.8 g氯化钠,加入5 mL乙酸乙酯,涡旋2 min,超声10 min,9 000 r/min离心6 min,取有机相于15 mL聚丙烯离心管。残渣再加入5 mL乙酸乙酯,涡旋2 min,9 000 r/min离心6 min,合并提取液。40 ℃水浴氮吹浓缩至近干,待水解。
脂肪提取法:参照GB 5009.6—2016《食品安全国家标准食品中脂肪的测定》[26]第三法(碱水解法)提取样品中的脂肪。之后准确称取0.1 g脂肪提取物(精确至0.1 mg)于15 mL聚丙烯离心管中,加入50 μL质量浓度为2 mg/L(以氯丙醇计)的酯混合内标工作液,混匀,待水解。
(2)水解
于待水解样中加入200 μL叔丁基甲基醚,涡旋混匀,加入200 μL甲醇钠-甲醇溶液,涡旋混匀,室温反应3 min,立即加入600μL酸化溴化钠溶液,涡旋30s,终止水解反应并中和过量的甲醇钠。加入1 mL正己烷,涡旋30 s,4 000 r/min离心2 min,弃去上层正己烷层,再加入1 mL正己烷重复一次,去除非极性杂质成分。水相中加入1 mL乙醚-乙酸乙酯溶液(6∶4,V/V),涡旋1 min,8 000 r/min离心2 min,取有机相于10 mL具塞密闭玻璃管中,水相中再加入1 mL乙醚-乙酸乙酯溶液(6∶4,V/V)重复提取一次,合并有机相,待衍生。
(3)衍生
七氟丁酰基咪唑衍生法:将待衍生液于40 ℃水浴氮吹浓缩至近干,加入1 mL异辛烷,加入50 μL七氟丁酰基咪唑,立即密塞,涡旋混合,70 ℃保温20 min。取出冷却至室温,加入2 mL饱和氯化钠溶液,涡旋1 min,静置分层。取异辛烷层,经无水硫酸钠脱水后,供气相色谱-质谱(gas chromatography-mass spectrometry,GC-MS)测定。
苯硼酸衍生法:向待衍生液中加入200 μL苯硼酸溶液,涡旋30 s,30 ℃反应20 min。衍生反应结束后,40 ℃水浴氮吹浓缩至近干。加入1 mL异辛烷复溶,上清液经0.22 μm滤膜过滤,供GC-MS测定。
1.3.3 仪器条件
(1)色谱条件
HP-5MS色谱柱(30 m×0.25 mm×0.25 μm);进样口温度:250 ℃;载气:氦气(He),纯度≥99.999%,流速:0.8 mL/min;进样量:1 μL;进样模式:不分流进样;视衍生方式不同分别采用不同的程序升温:柱温1(七氟丁酰基咪唑衍生物采用):50 ℃保持5 min,以2 ℃/min的速度升至80 ℃,300 ℃后运行5 min;柱温2(苯硼酸衍生物采用):50 ℃保持1 min,以10 ℃/min的速度升至210 ℃,300 ℃后运行5 min。
(2)质谱条件
电离方式:电子电离(electron ionization,EI)源,70 eV;离子源温度:300℃;传输线温度:280 ℃,四极杆温度:180 ℃;扫描方式:选择离子监测(selectedionmonitoring,SIM)模式。
1.3.4 数据处理
取一系列醇混合标准工作液加入醇混合内标工作液,衍生化处理后经GC-MS测定,绘制标准曲线,样品中含量采用内标法定量,本实验氯丙醇酯含量均以氯丙醇计(奶粉中)。用安捷伦MSD ChemStation软件对数据进行采集,使用Origin 8.0软件绘图。
2 结果与分析
2.1 质谱特征离子的考察
氯丙醇及其同位素内标经七氟丁酰基咪唑或苯硼酸衍生后,其衍生物按1.3.3(1)色谱条件通过HP-5MS毛细管色谱柱均可实现良好分离,经全扫描模式确认特征离子碎片。3-MCPD和2-MCPD七氟丁酰基咪唑衍生物在电子轰击下,C-O键断裂,脱去-O(CO)C3F7后形成m/z 289和291的离子,继续脱去-HCl形成m/z 253的离子,或脱去-CH2形成m/z 275和277的离子,或脱去-O(CO)C3F7后形成m/z 75和77的离子;3-MCPD和2-MCPD 苯硼酸衍生物的分子离子是m/z 196,同位素分子离子是m/z 195和198,3-MCPD苯硼酸衍生物失去-CH2Cl后形成了m/z 147和146的离子,2-MCPD苯硼酸衍生物失去-OCH2(CHCl)CH2后形成的m/z 104的离子;同位素内标与对应的氯丙醇质谱裂解机理相同。根据各离子碎片的响应强度和干扰情况,确定两种氯丙醇及内标衍生物的特征离子如表1所示。
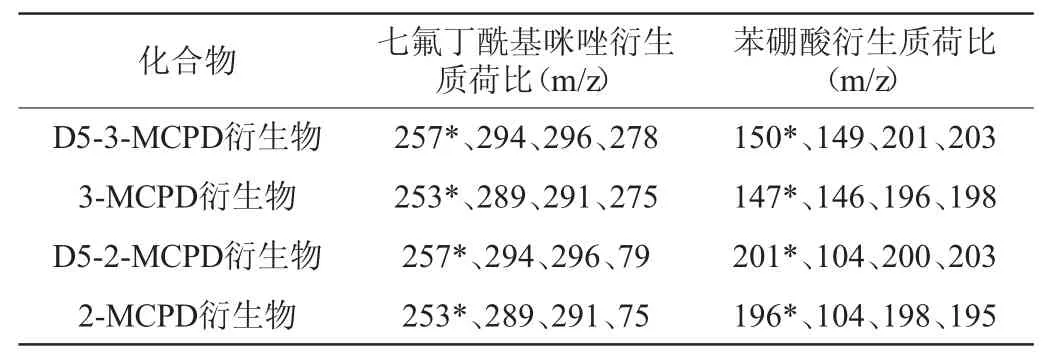
表1 氯丙醇及其同位素内标衍生物的特征离子参数Table 1 Characteristic ion parameters of chloropropanol and its isotope internal standard derivatives
2.2 衍生方法的选择
本研究以相同浓度的醇标样溶液,分别采用苯硼酸和七氟丁酰基咪唑两种衍生试剂处理,衍生后的样品在同一台仪器上按照相应的仪器条件进行采集,通过考察衍生物的色谱峰响应面积、对色谱柱的选择性及影响,比较了两种衍生方法的差异,结果见表2,4种MCPD衍生物的提取离子色谱图见图1。

表2 两种衍生方法的比较Table 2 Comparison of two derivative methods

图1 4种MCPD衍生物的提取离子色谱图Fig.1 Extraction ion chromatograms of four MCPD derivatives
由图1可知,同浓度醇标样溶液的2种衍生物在色谱行为上相比较,七氟丁酰基咪唑衍生物峰形明显好于苯硼酸衍生物,峰形更尖锐、对称,方法灵敏度更高。实验发现,苯硼酸衍生法的上机液容易造成仪器的污染,并且对色谱柱有很大的破坏力。采集约60针样品后就会出现明显的色谱峰拖尾、响应降低甚至不出峰、仪器背景快速升高、色谱柱柱效显著降低等问题,致使仪器日常维护频繁、色谱柱损伤严重、使用寿命极大缩短,工作效率大大降低。因此,本研究选用灵敏度高、耐用性好的七氟丁酰基咪唑衍生法。
2.3 直接提取法的条件优化
2.3.1 提取溶剂的选择
氯丙醇酯是脂溶性物质,分别选用乙酸乙酯、正己烷、甲基叔丁基醚、乙腈作为溶剂,提取后再经水解和衍生化处理,采用气相色谱-质谱测定分析。通过加标回收试验(加标水平0.10 mg/kg,以氯丙醇计),比较了提取溶剂对氯丙醇酯的选择性,结果见表3。

表3 不同溶剂对氯丙醇酯的回收率Table 3 Recovery rate of chloropropanol esters with different solvents
由表3可知,乙酸乙酯提取液对目标物的回收率均为98%以上;甲基叔丁基醚提取液回收率次之;正己烷提取时乳化严重,回收率偏低;乙腈提取时能同时沉淀蛋白,减小基质共萃出,但对氯丙醇酯的选择性较差,回收率最低。综合对比,本研究选用了毒性较小、选择性好的乙酸乙酯为提取溶剂。
2.3.2 碱水解试剂用量和水解时间优化
在碱水解过程中,氯丙醇酯在甲醇钠作用下发生酯键断裂生成氯丙醇,甲醇钠用量过多或水解时间过长都会引起氯丙醇的降解,导致方法灵敏度的降低[20]。实验以氯丙醇酯检出阳性的婴幼儿配方乳粉样品为研究对象,按照直接提取法进行样品提取得待水解样,于待水解样中加入200 μL叔丁基甲基醚,涡旋混匀,待水解,分别设置不同的水解试剂用量和水解时间对实验条件进行优化。
于加入200 μL叔丁基甲基醚的待水解液中加入甲醇钠-甲醇溶液,设置甲醇钠-甲醇溶液用量分别为50 μL、100 μL、200 μL、500 μL、1 000 μL,水解时间为3 min。水解结束后采用七氟丁酰基咪唑衍生,GC-MS分析,分别考察水解产物氯丙醇定量离子色谱峰响应面积的变化,每个条件分别做3次平行,结果见图2A。由图2A可知,实验发现MCPD及其内标衍生物响应面积随着碱水解试剂用量的增多逐渐升高,在试剂用量为200 μL时,MCPD及其内标衍生物响应面积到达峰值,而后逐渐降低后保持平缓,为了提高实验灵敏度,便于准确定量,所以选择最佳甲醇钠-甲醇溶液用量为200 μL。
于待水解液中,加入200 μL甲醇钠-甲醇溶液,水解时间分别设定为0.5 min、1.0 min、2.0 min、3.0 min、4.0 min、6.0 min、10.0 min,水解结束后采用七氟丁酰基咪唑衍生进行GC-MS分析,分别考察水解产物氯丙醇定量离子色谱峰响应面积的变化,每个条件分别做3次平行,结果见图2B。由图2B可知,MCPD及其内标衍生物响应面积随着水解时间的延长,先逐渐升高,在水解时间3 min时到达峰值后又逐渐降低,所以选择水解时间3 min为最优条件。

图2 碱水解试剂用量(A)及水解时间(B)对MCPD衍生物响应的影响Fig.2 Effect of alkali hydrolysis reagent dosage (A) and hydrolysis time (B) on MCPD derivatives response
2.4 直接提取法的方法学研究
2.4.1 线性范围和定量限
分别移取5 μL、10 μL、20 μL、50 μL、100 μL、200 μL质量浓度为2 mg/L的醇混合标准工作液,加入2 mg/L醇混合内标工作液50 μL,将标准系列进行七氟丁酰基咪唑衍生化处理后,按相应仪器条件测定分析。以氯丙醇衍生物与其对应内标衍生物的定量离子响应面积之比为纵坐标,质量浓度比为横坐标,进行线性回归,得到线性回归方程及相关系数,线性范围、线性方程及相关系数见表4。
由表4可知,目标化合物线性关系良好,相关系数均大于0.999。在空白婴儿配方乳粉基质中添加氯丙醇酯标准溶液,添加水平为0.010 mg/kg、0.020 mg/kg、0.050 mg/kg(以氯丙醇计),按照直接提取法进行处理,以3倍、10倍信噪比对应的含量分别为检出限、定量限,确定了方法检出限、定量限分别为0.003 mg/kg、0.010 mg/kg。

表4 目标化合物的线性方程、相关系数、检出限和定量限Table 4 Linear equation,correlation coefficient,LODs and LOQs of target compounds
2.4.2 方法的准确度
在空白婴儿配方乳粉基质中分别添加氯丙醇酯标准溶液,添加水平为0.01 mg/kg、0.10 mg/kg和0.20 mg/kg(以氯丙醇计),按直接提取法进行处理,每个加标水平做6个平行,计算回收率和相对标准偏差(relative standard deviation,RSD),结果见表5。结果表明,3个浓度水平加标的平均回收率在95.8%~115.7%之间,回收率试验结果相对标准偏差为2.8%~4.6%。结果表明本方法准确度较好,能够满足婴儿配方乳粉中氯丙醇酯定量分析的需要。

表5 目标物在婴幼儿配方乳粉中的加标回收率试验结果(n=6)Table 5 Results of adding standard recovery tests of target compounds in infant formula (n=6)
2.4.3 方法的精密度
选取1份婴幼儿配方乳粉阳性样品,分别称取6份样品,按直接提取法进行处理,测定6个平行中氯丙醇酯的含量(以醇计),计算相对标准偏差(RSD),结果见表6。由表6可知,2种氯丙醇酯的RSD分别为1.9%、2.2%,均<5%,结果表明本方法测定结果稳定、精密度良好,能够满足婴儿配方乳粉中氯丙醇酯含量的检测要求。

表6 方法的精密度试验结果(n=6)Table 6 Results of precision tests of the method (n=6)
2.5 两种提取方法的比较
本研究选用3份婴幼儿配方乳粉阳性样品和1份英国FAPAS(Food Analysis Performance Assessment Scheme)质控样(编号:T2661QC,基质为婴儿配方乳粉,3-MCPDE、2-MCPDE的理论含量分别为0.050 2 mg/kg、0.019 0 mg/kg,以醇计)。按照样品前处理方法分别进行试验,分别采用直接提取法和提取脂肪法,比较提取方法对氯丙醇酯定量结果的影响,平行测定6次,结果见表7。由表7可知,两种方法的测定结果无明显差别,可以进行相互验证。

表7 提取方法对氯丙醇酯定量结果的影响(n=6)Table 7 Effect of extraction methods on the quantitative results of chloropropanol esters (n=6)
3 结论
通过提取方式及衍生方法的选择、提取溶剂、碱水解试剂用量和水解时间的优化等多方面实验,本研究建立了气相色谱-质谱法测定婴幼儿配方乳粉中3-MCPDE和2-MCPDE含量的分析方法。实验发现,2种氯丙醇酯在一定范围内线性关系良好、准确度和精密度较高;七氟丁酰基咪唑衍生法比苯硼酸衍生法具有较好的耐用性和更高的灵敏度;直接提取法和提取脂肪法两种方法定量结果差异不大,可以进行相互验证,但直接提取法步骤简便、高效、快速,可大大提高工作效率。综上所述,本方法能够实现婴幼儿配方乳粉中氯丙醇酯的快速批量测定,可以为婴幼儿配方乳粉中氯丙醇酯的风险监测和暴露评估提供技术支持。
猜你喜欢
食品科学(2023年4期)2023-03-06 12:49:32
分子催化(2022年1期)2022-11-02 07:10:30
食品工业科技(2021年23期)2021-12-16 02:21:50
食品安全导刊(2021年21期)2021-08-30 08:21:58
中国乳业(2020年12期)2020-04-12 01:12:46
食品与生活(2017年5期)2017-05-27 20:28:38
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
当代化工研究(2016年2期)2016-03-20 16:21:23
中国洗涤用品工业(2016年2期)2016-02-28 19:03:17
合成化学(2015年10期)2016-01-17 08:56:48
