
微波消解-电感耦合等离子体质谱法测定高岭土中10种微量元素
2022-03-10 12:34:22李丽君薛静
岩矿测试 2022年1期
李丽君, 薛静
(中国地质调查局沈阳地质调查中心, 自然资源部黑土地演化与生态效应重点实验室,
高岭土,化学式为Al4[Si4O8](HO)8,由铝硅酸盐类矿物经长期风化而成或热液蚀变或表生沉积作用形成的黏土矿物。高岭土致密的晶质结构决定了其用途非常广泛,常见于陶瓷、造纸、颜料、橡胶、催化剂、净水剂[1-3]及其他含铝产品制造行业。高岭土中K2O、Na2O等主量组分影响着产品的主体性能,而As、Sb等微量杂质元素的含量则对高岭土相关产品卫生指标产生严重影响[4-5],进而影响高岭土的矿产资源综合评价和利用。国家标准《高岭土及其试验方法》(GB/T 14563—2008)主要集中于主量元素以及SO3和烧失量的测定,对于稀土元素、As、Sb等微量元素的测定则没有涉及。高岭土的三种国家标准物质GBW03121、GBW03122、GBW03122a中均未提供As、Sb等微量元素的标准值,制约了高岭土的应用。因此,高岭土中微量元素的准确测定具有十分重要的意义。
地质样品中As、Sb等微量元素测试方法主要有原子荧光光谱法(AFS)[6-8]、电感耦合等离子体发射光谱法(ICP-OES)[9-10]、X射线荧光光谱法(XRF)[11-12]、电感耦合等离子体质谱法(ICP-MS)[13-15]。刘佩佩[7]、赵小学等[8]采用AFS法测定土壤和水系沉积物中5种还原性元素及砷的形态。袁静等[12]采用波长色散与能量色散X射线荧光光谱法对比测定了地质样品中的部分主微量元素,但是并未给出具体微量元素的方法检出限及测定下限。ICP-MS法[13-14]因其灵敏度高、动态线性范围宽、多元素同时测定的优势,在地质样品和生物样品中的微量、痕量、超痕量元素测定方面均得以广泛应用[15]。该方法因样品类型复杂、基体效应较强[14],对样品的溶样方法及干扰消除提出了更高要求。黄冬根等[16]采用敞开二酸体系,ICP-MS法测定了高岭土中6种主微量元素;罗勉等[17]采用敞开三酸体系,ICP-OES法测定高岭土中8种元素;马生凤等[18]采用敞开四酸溶样ICP-OES测定了硫化物矿石中22种元素;张亚峰等[19]采用敞开五酸体系测定地质样品中46种元素;张晨芳等[20]采用密闭压力酸溶ICP-MS法测定岩浆岩中稀有元素;王佳翰等[21]采用碱熔ICP-MS法测定海洋沉积物中48种元素。以上方法各有特点,敞开消解法的优势在于消解时间较高压密闭法短;高压密闭法[20]的检出限较敞开酸溶法更低;XRF法[11-12]的前处理过程相对简便。上述溶样方法共同的缺点在于样品前处理效率与各元素足够低的检出限不能同时兼顾。高温碱熔法[21]测定的元素虽然较多,但前处理过程复杂,基体干扰严重。微波消解法[22-23]操作简便、消解时间短,可防止样品污染及挥发性组分损失,结合ICP-MS法能有效弥补该不足。
本文通过比较硝酸-氢氟酸二酸、硝酸-氢氟酸-过氧化氢三酸两种溶样体系,探讨了微波消解的程序升温模式和恒温模式对元素回收率的影响,考察了动能歧视模式(KED)对多原子离子质谱干扰的消除效果,用岩石标准物质的制备溶液直接配制标准曲线,测定中在线加入103Rh内标,克服了质谱和非质谱干扰,建立了采用微波消解ICP-MS法测定高岭土样品中As、Sb、Bi、Cd、Cr、Cu、Mo、Ni、Pb、Zn等10种微量元素的分析方法。
1 实验部分
1.1 仪器及工作条件
iCAP-RQ型电感耦合等离子体质谱仪(KED模式,美国ThermoFisher公司);MARS classic微波消解仪(美国CEM公司);超纯水器(重庆市奥凯龙公司); BSA1245-CW 型电子天平(德国Sartorius公司);DHG-9145A型电热恒温鼓风干燥箱(上海齐欣科学仪器有限公司);EG-20A型数显控温电热板(美国LabTech公司);高纯氦气;高纯氩气。
微波消解仪的工作参数见表1。
表1微波消解仪的工作参数
Table 1 Working parameters of microwave digestion apparatus
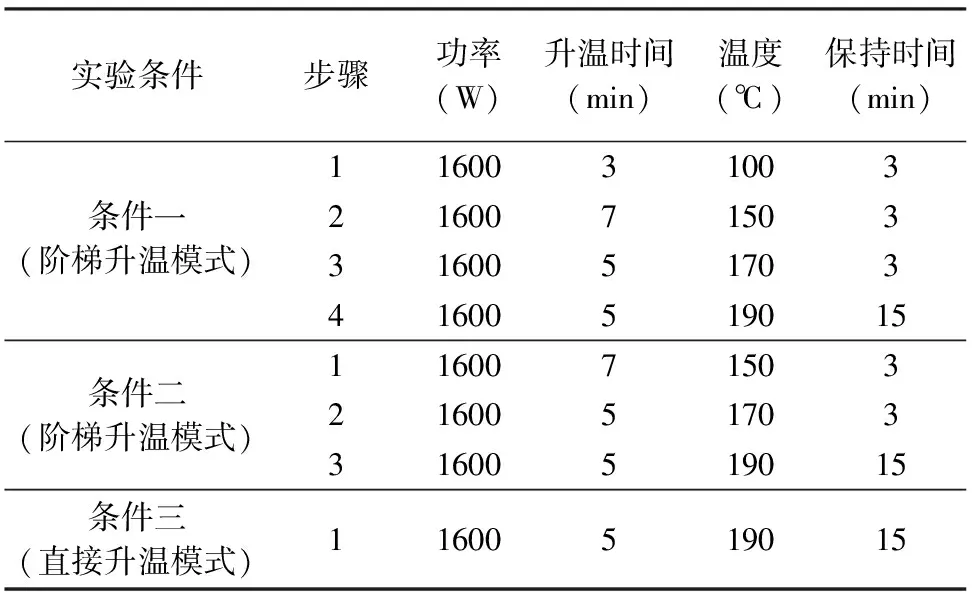
实验条件步骤功率(W)升温时间(min)温度(℃)保持时间(min)1160031003条件一(阶梯升温模式)21600715033160051703416005190151160071503条件二(阶梯升温模式)216005170331600519015条件三(直接升温模式)11600519015
ICP-MS仪器的工作参数为仪器全自动优化调谐给出,仪器灵敏度、背景值、稳定性调至最佳。入射功率1250W;扫描方式: 跳峰;雾化气流速0.60~0.80L/min;冷却气流速13L/min;辅助气流速0.7L/min;积分时间0.5s;分辨率100;采样深度100step;半导体冷却温度2~ 4℃;碰撞气(氦气)流速3.2mL/min;具有动能歧视模式。
1.2 标准溶液和主要试剂
103Rh标准储备溶液(1000μg/mL)。As、Sb等10种微量元素的标准溶液(均购自国家有色金属及电子材料分析测试中心)。
103Rh标准工作溶液(5.0μg/L):用103Rh标准储备溶液逐级释配制而成。
调谐液(购自美国 ThermoFisher 公司)。硝酸、氢氟酸、高氯酸、盐酸、浓硫酸均为优级纯试剂。
实验用水均为超纯水。
1.3 标准物质
在地质类样品测试中使用的标准物质,应尽可能选用与待测样品的特性相似或相同矿种的标准物质来进行质量监控,尽可能地降低由粒度效应、矿物效应等不均匀性效应引起的系统误差和偶然误差。由于国家高岭土化学成分标准物质GBW03121、GBW03122、GBW03122a均没有对本方法提到的As、Sb等10种元素的标准值,因此选用岩石国家标准物质GBW07103、GBW07104、GBW07106 和GBW07107、GBW07109、GBW07110、GBW07111和GBW07113(中国地质科学院地球物理地球化学勘查研究所研制)进行标准曲线的配制、方法验证及不确定度评估。
1.4 样品处理
将岩石标准物质GBW07103、GBW07104、GBW07106、GBW07107、GBW07109、GBW07110、GBW07111、GBW07113于105~110℃烘2h,并在干燥器中冷却至室温后称量。
高岭土样品为不同两地高岭土样品,经过破碎、缩分等工序制成粒度为200目的样品,在干燥器内保存备用。
准确称取样品50mg(精确至0.0001g)于33mL聚四氟乙烯消解罐中,用几滴超纯水润湿后,加入2mL硝酸、5mL氢氟酸、1mL过氧化氢,将聚四氟乙烯消解管放入微波消解仪的支架上,采用表1的微波消解程序升温条件二进行消解,程序结束后,将消解管中溶液转移至聚四氟乙烯坩埚中,在150℃电热板上加热至溶液体积近干,趁热加入1.5mL硝酸溶解盐分,用超纯水冲洗坩埚杯壁转移至50mL容量瓶中定容至刻度,摇匀静置待测。配制内标103Rh标准溶液,质量浓度为5.0μg/L,采用在线内标方式引入。
1.5 标准曲线的制备
本方法选择样品空白溶液、国家标准物质GBW07103、GBW07104、GBW07106 和GBW07107作为曲线点,按照样品微波消解的溶样方法,同一批次进行处理,利用测得的各元素浓度与响应信号的关系来配制标准曲线,用于实际样品的定量分析。由于标准曲线点和实际样品溶液的基质是一致的,消除了因操作过程、黏度不同对溶液的传输效率的影响,从而确保基质对待测元素的化学影响基本一致,进而使非质谱干扰得到了扣除,同时实验操作过程及溶液的黏度、表面张力、密度和蒸汽压等因素引起的物理影响趋于一致[24]。标准曲线用标准物质GBW07103、GBW07104、GBW07106 和GBW07107的样品制备过程与待测样品的溶样过程相同。以元素响应值和内标响应值的比值为纵坐标,标准物质的含量为横坐标绘制标准曲线。
2 结果与讨论
2.1 酸溶体系的选择
地质样品中用来溶样的酸主要包括盐酸、硝酸、硫酸及高氯酸。荀颖怡[25]、张亚峰等[19]通过四酸和五酸体系溶样方法测定地质样品中的元素。高氯酸作为强氧化剂,沸点较低,加热过程中如遇到无机还原剂会因反应剧烈引起爆炸。硫酸的沸点高,能够将氢氟酸与高岭土中的二氧化硅反应生成的氟化物以及As、Sb等易还原元素通过升温而挥发,在密闭的消解管中容易造成管内局部压力过高引起溢出。盐酸因其在质谱测定时引入多原子离子的质谱干扰,故在ICP-MS测定中应用较少。作为微波消解系统常用的过氧化氢则具有明显的优势。王娜等[22]通过加入过氧化氢提高硝酸的氧化能力,使得样品中的有机质消解完全。本实验采用微波消解装置,以标准物质GBW07109作为样品,考察了硝酸-氢氟酸体系、硝酸-氢氟酸-过氧化氢体系对10种待测元素回收率的影响,结果表明采用硝酸-氢氟酸-过氧化氢三酸体系10种待测元素的测定值和标准值的相对误差较小,As、Sb的回收率明显高于硝酸-氢氟酸二酸体系,其余元素的回收率则较为接近。说明溶样体系中加入过氧化氢能够提高待测元素的回收率,本实验中由于高岭土的晶质致密结构,为确保样品能够完全消解,选择硝酸-氢氟酸-过氧化氢三酸体系作为溶样体系,其中硝酸、氢氟酸、过氧化氢用量分别为2mL、5mL、1mL。
2.2 微波消解升温模式的优化
微波消解的升温模式对样品的提取效率有着直接影响。王娜等[22]对比了封闭压力酸溶和微波消解方式对测定值的影响,结果表明采取微波消解溶样,用较少的时间也能达到较好的测定结果;王佩佩等[23]采用微波五阶升温模式酸溶消解地质样品中的稀土元素,消解时间115min;吴佳伦等[26]采用四阶升温模式酸溶消解土壤样品中12种重金属元素,消解时间60min;张祎玮等[27]采用二阶升温模式酸溶消解土壤样品中稀土元素,消解时间40min,均得到了较好的测试效果。本文对比了阶梯升温和直接升温消解模式对标准物质GBW07109中10种待测元素回收率的影响,结果如图1所示。

图1 消解条件对元素回收率的影响Fig.1 Effect of digestion conditions on recovery of elements
由图1可知,当采用阶梯升温模式(条件一、条件二)时,消解酸溶液能够充分渗入样品晶格内部,消解完全,待测元素回收率高;条件三则为直接升温模式,样品溶液从室温直接加热到设置的最高温度,消解溶剂浸润的时间短,待测元素的回收率偏低,同时直接升温反应剧烈导致消解管压力骤然升高。对比四阶升温和三阶升温模式对元素回收率的影响,不考虑消解溶液冷却的时间,三阶升温和四阶升温模式消解条件下,元素的回收率接近,表明在两种升温模式下,样品中各元素均提取完全,在三阶升温模式下,微波消解时间38min,低于张祎玮等[27]的消解时间,表明本方法的消解效率高。考虑到时间和效率的因素,在消解完全的前提下,本文的微波消解条件选择三阶升温模式,微波消解时间38min。
2.3 ICP-MS的干扰及消除
2.3.1非质谱干扰及消除
ICP-MS的非质谱干扰主要来源于样品溶液的基体效应,实验中得到Si、Al的溶解性固体总量太高则导致质谱信号的偏移,通过对待测元素信号的抑制作用影响测定结果的精密度和准确度。本实验通过控制样品的稀释因子在500~1000之间,以减轻基体干扰程度,使得待测样品溶液可溶解固体总量在0.2%以下[28]。为监测和校正信号的漂移及基体效应,本实验通过采用在线加入内标103Rh对待测元素的信号值进行补偿。样品处理过程[24]采取通过选择岩石标准物质的溶液浓度直接绘制标准曲线,实际测定样品的溶液和样品空白溶液的酸溶过程以及上机仪器操作过程相对一致,确保了操作误差为系统误差从而消除基体效应的干扰。
2.3.2质谱干扰及消除
质谱干扰通常包括同质异位素重叠干扰、多原子离子干扰[29]、难熔氧化物干扰、双电荷离子干扰等四种质谱干扰。本实验由于待测元素较多、质量数较大,主要表现为多原子离子干扰。王佳翰等[21]、徐进力等[30]采用动能歧视模式(KED)有效地降低了氧化物干扰,提高信背比。赵志飞等[31]采用氧气反应模式降低了多原子离子干扰。动能歧视模式(KED)是在ICP- MS的碰撞池中引入He 与多原子干扰离子发生碰撞,从而降低质谱千扰。本文考察了ICP-MS的测定模式中标准模式(STD)及动能歧视模式(KED)对测定结果的影响,结果表明采用带动能歧视模式(KED)的ICP-MS法能够有效地降低多原子离子和难熔氧化物干扰,方法检出限更低,灵敏度更高。
2.4 分析方法评价
2.4.1方法准确度和精密度
采用国家标准物质进行方法性能验证,按照图1中确定的微波消解条件二,选取GBW07109、GBW07110、GBW07111和GBW07113四个标准物质按照本方法进行测定,平行测定5次。从表2测定结果可以看出GBW07109、GBW07110、GBW07111和GBW07113各元素的相对误差均在-9.1%~3.2%之间,同时小于标准物质的分析结果相对误差允许限允许限(YB),元素回收率在90.9%~103.2%之间,相对标准偏差(RSD)在1.2%~5.8%之间,均符合《地质矿产实验室测试质量管理规范》(DZ/T 0130—2006)要求。
表2方法准确度和精密度(n=5)
Table 2 Accuracy and precision tests of the method (n=5)
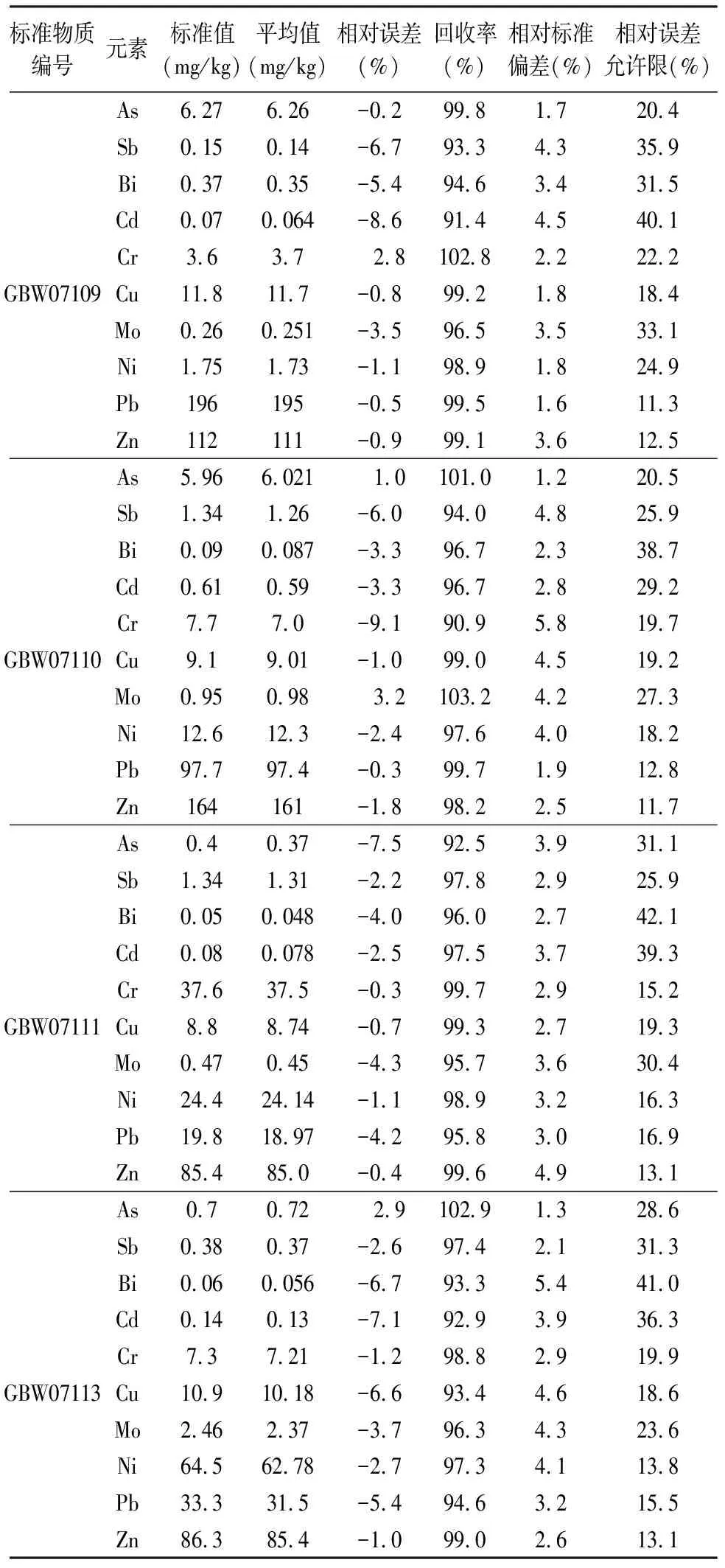
标准物质编号元素标准值(mg/kg)平均值(mg/kg)相对误差(%)回收率(%)相对标准偏差(%)相对误差允许限(%)As6.276.26-0.299.81.720.4Sb0.150.14-6.793.34.335.9Bi0.370.35-5.494.63.431.5Cd0.070.064-8.691.44.540.1Cr3.63.7 2.8102.82.222.2GBW07109Cu11.811.7-0.899.21.818.4Mo0.260.251-3.596.53.533.1Ni1.751.73-1.198.91.824.9Pb196195-0.599.51.611.3Zn112111-0.999.13.612.5As5.966.021 1.0101.01.220.5Sb1.341.26-6.094.04.825.9Bi0.090.087-3.396.72.338.7Cd0.610.59-3.396.72.829.2Cr7.77.0-9.190.95.819.7GBW07110Cu9.19.01-1.099.04.519.2Mo0.950.98 3.2103.24.227.3Ni12.612.3-2.497.64.018.2Pb97.797.4-0.399.71.912.8Zn164161-1.898.22.511.7As0.40.37-7.592.53.931.1Sb1.341.31-2.297.82.925.9Bi0.050.048-4.096.02.742.1Cd0.080.078-2.597.53.739.3Cr37.637.5-0.399.72.915.2GBW07111Cu8.88.74-0.799.32.719.3Mo0.470.45-4.395.73.630.4Ni24.424.14-1.198.93.216.3Pb19.818.97-4.295.83.016.9Zn85.485.0-0.499.64.913.1As0.70.72 2.9102.91.328.6Sb0.380.37-2.697.42.131.3Bi0.060.056-6.793.35.441.0Cd0.140.13-7.192.93.936.3Cr7.37.21-1.298.82.919.9GBW07113Cu10.910.18-6.693.44.618.6Mo2.462.37-3.796.34.323.6Ni64.562.78-2.797.34.113.8Pb33.331.5-5.494.63.215.5Zn86.385.4-1.099.02.613.1
2.4.2方法检出限及测定下限
本方法在仪器最佳化条件下,按照样品处理步骤得到的全流程样品空白溶液,平行12次测定,以测定结果的3倍的标准偏差为检出限计算,10倍的标准偏差为测定下限,考虑稀释因子1000,方法检出限、测定下限结果见表3。从表3可以看出,采用微波消解提取,ICP-MS测定,本方法检出限在0.01~0.09mg/kg之间,测定下限在0.03~0.30mg/kg之间,方法检出限和测定下限均小于《铝土矿石分析方法》(YS/T 575—2007)、《硅酸盐分析方法》(GB/T 14506—2010)中相关元素方法检出限的要求,与李诚等[32]测定金刚石中次量元素的方法检出限相比则更低,同时也低于《区域地球化学样品分析方法》(DZ/T 0279—2016)规定的方法检出限,而As、Sb、Bi的方法检出限则高于《土壤和沉积物 汞、砷、硒、铋、锑的测定 微波消解/原子荧光法》(HJ 680—2013)中对应元素的检出限,表明无机元素在岩石致密晶格及疏松土壤微粒的赋存状态不同,岩石基体不如土壤与酸类及氧化剂的反应充分,灵敏度相对偏低。本实验中的方法检出限满足地质行业中岩石样品测定的要求。
表3方法的方法检出限及测定下限(n=12)
Table 3 Detection limits and lower limit of the method (n=12)
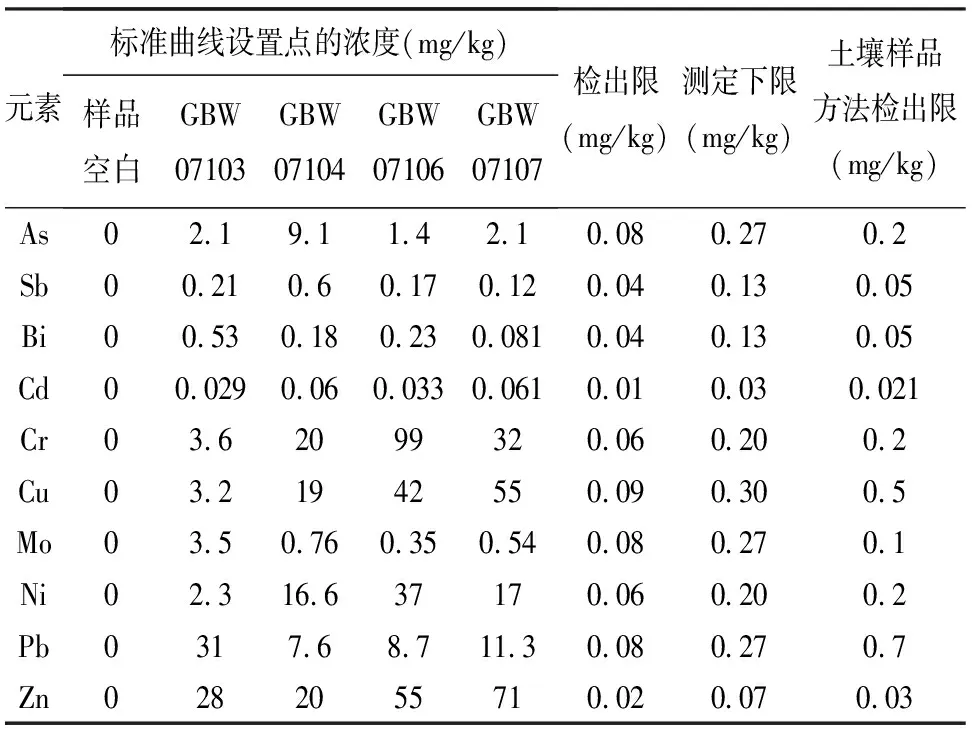
元素标准曲线设置点的浓度(mg/kg)样品空白GBW07103GBW07104GBW07106GBW07107检出限(mg/kg)测定下限(mg/kg)土壤样品方法检出限(mg/kg)As02.19.11.42.10.080.270.2Sb00.210.60.170.120.040.130.05Bi00.530.180.230.0810.040.130.05Cd00.0290.060.0330.0610.010.030.021Cr03.62099320.060.200.2Cu03.21942550.090.300.5Mo03.50.760.350.540.080.270.1Ni02.316.637170.060.200.2Pb0317.68.711.30.080.270.7Zn0282055710.020.070.03
注:“土壤样品方法检出限”为DZ/T 0279—2016中数据。
3 分析方法的不确定度评估
为了对方法的准确度进行精确评估,本研究根据关于不确定度的定义,计算本分析方法的A类不确定度。同时根据国家标准 《测定不确定度评定和表示》(GB 27418—2017)及欧盟和国际上分析方法不确定度的评估[12,33]方法,计算方法的扩展不确定度。
3.1 A类不确定度的计算

3.2 扩展不确定度的计算
根据国家标准的定义,由在一个测量模型中各输入量的标准测量不确定度获得的输出量的标准测量不确定度为合成标准不确定度,而合成标准测量不确定度与一个大于1的数字因子的乘积计算得到的不确定度则为扩展不确定度UΔ。本方法在 95%置信水平下(k=2),扩展不确定度UΔ为2倍的标准不确定度(A类不确定度)。
通过计算得到了测定平均值和标准值的绝对差Δm、A类不确定度Δx以及方法的扩展不确定度UΔ,数据列于表4。
根据欧盟和国际上对分析方法不确定度的评价标准,当测定平均值和标准值的绝对差Δm<扩展不确定度UΔ时,表示测量值和标准值之间不存在显著性差异。由表4数据可见,选定样本中 10种微量元素的测量值与标准值之间无显著性差异,表明采用本方法测定各元素的含量与标准值相符,方法性能是有效、可靠的。
4 实际样品分析
分析时称取两个地区高岭土的实际样品各50mg,并采用标准加入法对测定结果进行质量监控,向每个样品中加入一定量的混合标准溶液,使得样品中各元素的加标量均为50mg/kg,每个样品在本方法确定的最佳消解方法及ICP-MS检测条件下重复测定3次,测定结果及回收率、RSD结果见表5所示,可以看出两个样品中10种元素的加标平均回收率在94.6%~98.5%之间,RSD在2.9%~7.1%,符合《地质矿产实验室测试质量管理规范》(DZ/T 0130—2006)回收率(90%~110%)的要求,表明本方法适合于高岭土实际样品的分析。
表4分析方法的不确定度计算结果
Table 4 Calculation results of uncertainty of the method

标准物质编号元素标准值(mg/kg)标准值的不确定度(mg/kg)A类不确定度Δx(mg/kg)扩展不确定度UΔ(mg/kg)测定平均值和标准值的绝对差Δm(mg/kg)As6.270.90.10.20.01Bi0.370.070.020.040.02Cd0.070.020.0040.0080.01Cr3.61.60.10.20.1GBW07109Cu11.81.70.30.60.1Mo0.260.090.010.020.01Ni1.750.720.040.080.02Pb19620481Sb0.150.060.010.020.01Zn1127361As5.960.890.090.180.061Bi0.090.040.0020.0040.003Cd0.610.130.020.040.02Cr7.72.20.51.00.7GBW07110Cu9.11.50.51.00.1Mo0.950.130.050.100.03Ni12.61.90.61.20.3Pb97.79.32.34.60.3Sb1.340.210.080.160.08Zn164105103As0.40.30.020.040.03Bi0.050.030.0020.0040.002Cd0.080.030.0040.0080.002Cr37.63.41.42.80.1GBW07111Cu8.81.50.30.60.1Mo0.470.40.020.040.02Ni24.42.30.91.80.3Pb19.82.30.71.40.8Sb1.340.210.050.100.03Zn85.49.44.28.40.4As0.70.30.010.020.02Bi0.060.020.0040.0080.004Cd0.140.030.0060.0120.01Cr7.33.30.30.60.1GBW07113Cu10.91.60.61.20.7Mo2.460.260.130.260.09Ni64.58361.7Pb33.33.11.22.41.8Sb0.380.050.010.020.01Zn86.37.82.85.60.9
表5高岭土实际样品的测定结果(n=3)
Table 5 Analytical results of the real kaolin samples (n=3)

元素样品1样品2测定值(mg/kg)加标后测定值(mg/kg)平均回收率(%)相对标准偏差(%)测定值(mg/kg)加标后测定值(mg/kg)平均回收率(%)相对标准偏差(%)As51.24100.298.05.241.490.698.54.2Sb5.8154.797.85.48.057.198.34.2Bi1.5450.597.94.61.550.798.53.8Cd2.2051.297.93.83.652.898.43.5Cr8.7057.597.76.89.758.898.24.6Cu30.078.496.85.136.184.897.44.1Mo5.2354.197.84.96.956.098.33.7Ni1.0550.098.03.81.250.498.52.9Pb75.1122.695.06.255.1103.596.85.1Zn85.12132.494.67.195.5143.395.63.9
5 结论
建立了一种采用硝酸-氢氟酸-过氧化氢三酸体系,三阶升温模式微波消解样品,利用岩石标准物质制备的溶液直接配制标准曲线,采用动能歧视模式(KED),ICP-MS法测定高岭土中As、Sb等10种微量元素的分析方法。方法性能测试及不确定度的评估验证了方法准确、可靠。本方法称样量50mg,酸的总用量为8mL,消解时间为38min,能用于高岭土样品中As、Sb等10种微量元素的快速批量分析,为高岭土国家标准物质GBW03121、GBW03122、GBW03122a中As、Sb等10种微量元素的测定提供了借鉴。
本方法采用标准物质的溶液直接制备标准曲线,可以极大程度上消除基体干扰,但由于选用的标准物质较少,个别元素的线性范围较窄,对元素含量较高的样品具有一定的局限性,可根据需要选择元素含量较高的标准物质,扩大方法的适用范围,此项工作需进一步探讨。
致谢:论文审稿过程中匿名专家提出建设性的修改意见,在此致以诚挚的谢意。
猜你喜欢
保健与生活(2023年6期)2023-03-17 08:39:54
化工设计通讯(2022年10期)2022-12-31 20:42:50
波谱学杂志(2022年2期)2022-06-14 09:52:02
山东冶金(2019年1期)2019-03-30 01:35:02
红领巾·探索(2018年12期)2018-01-26 12:34:14
金色年华(2017年12期)2017-07-18 11:11:20
天津城建大学学报(2015年5期)2015-12-09 01:26:53
中国塑料(2015年11期)2015-10-14 01:14:14
橡胶工业(2015年7期)2015-08-29 06:33:06
现代检验医学杂志(2015年1期)2015-02-06 01:59:14
