
表面活性肽在大肠杆菌中的异源表达
2022-02-23 13:33:14孟淑娟李慧男庞正军慕静王凤寰
食品与发酵工业 2022年3期
孟淑娟,李慧男,庞正军,慕静,王凤寰
(北京工商大学 轻工科学技术学院,北京,100048)
表面活性肽是模拟天然脂质分子的结构特征自主设计的一类具有表面活性的小分子短肽,一般由4~8个疏水性氨基酸形成的尾部和1~2个亲水性氨基酸形成的头部构成。与其他化学表面活性剂相比,表面活性肽具有对环境无污染、易生物降解、抑菌性强、营养性等优势[1],符合现代绿色工业发展理念,在清洁、食品、化妆品、医药等多个领域都有广泛的应用。特别是在食品方面,表面活性肽通过其本身的特性,在食品生产过程中充当乳化剂、湿润剂、稳定剂等,提高了食品的口感和新鲜度[2]。ANJUM等[3]发现,Bacillussp.MTCC 5877所产的生物表面活性剂能减少生物被膜的形成从而有效洗脱蔬菜表面粘附的有害微生物及残留的重金属。
目前表面活性肽的制备方法采用最多的是通过酶解把蛋白质水解到一定程度,使水解液中含有一定量的活性肽[2],此类方法主要用于制备天然的生物活性肽。对于已知氨基酸序列的表面活性肽还可采取定向合成法,包括化学合成、酶法合成和DNA重组技术等[4]。MERRIFIELD[5]建立的固相合成法奠定了多肽固相合成的基础,其原理是:将目标多肽的C端羧基以共价键形式结合在高分子树脂上,氨基酸在此基体内合成。以此作为起始端,目的肽所需的氨基酸顺序依次进行缩合,通过肽键连接不断延长肽链直至合成完成。但化学合成表面活性肽过程复杂、成本高、收率低,而生物发酵的方式对环境友好,操作过程无需使用大量有毒试剂,且一旦发酵流程和条件确定后可重复性高,能够实现放大生产。因此对具有应用价值的肽的合成,该方法是极佳的选择。VAN HELL等[6]设计了可以自组装成囊泡结构的两亲性寡肽Ac-A-A-V-V-L-L-L-W-E2/7 —COOH,保持疏水尾长度不变,通过改变E的个数,使亲水基部分长度增加,从而使亲水基团与疏水基团的长度之比发生改变,使肽链的亲水性发生变化。他们使用大肠细菌对肽进行重组生产,作为固相合成的替代方法,所得产物得率高且生产过程较为简单。ZHOU等[7]通过大肠杆菌重组表达并纯化出了由35个氨基酸残基组成的抗菌肽CM4,最终得到纯度为98%的1.4 mg/L重组抗菌肽CM4,且其抗菌活性与化学合成的CM4相近。这些研究都表明生物发酵的方式能够代替化学合成成为多肽的新制备方法,因此本研究尝试通过基因重组和生物发酵诱导表达得到表面活性肽。
VON MALTZAHN等[8]首先报道了两端封闭型表面活性肽A6K(Ac-AAAAAAK-NH2)在水溶液中的自组装行为,并且提出其在表面活性剂、生物技术和纳米技术中均有潜在应用。在随后的研究中,人们发现A6K可以在水中自组装形成囊泡,因此在疏水性药物的包裹和输送中具有良好的应用前景[9-10]。本实验室前期通过化学方法设计合成了非封闭端表面活性肽——A6K,并对其进行了表面张力、自组装性能等的研究,发现其具有较好的应用前景[11]。本研究旨在通过生物发酵方法制备此种表面活性肽,通过将编码A6K的重复DNA片段(A6K)15克隆到4种不同的原核表达载体上,构建重组质粒,转化3种不同的表达宿主,选择最优载体和最优宿主后优化表达条件。经纯化和十二烷基硫酸钠-聚丙烯酰氨凝胶电泳(sodium dodecyl sulfate-polyacrylamide gel electrophoresis,SDS-PAGE)及Western blot鉴定,最终确定了适合此类表面活性肽表达的最优载体和最优宿主,并且通过优化表达条件获得了表达量较高的新型小分子多肽聚合物(A6K)15。
1 材料与方法
1.1 实验材料
1.1.1 菌种和质粒
质粒pET-28a(+)、pET-28a-CLCⅠ,实验室保存;质粒pET-28a-SUMO、pT7-GST-His、大肠杆菌(E.coli)克隆宿主DH5α、表达宿主BL21(DE3)、BL21(DE3) plysS、Rosetta(DE3),北京庄盟国际生物科技有限公司。
1.1.2 试剂和材料
限制性核酸内切酶BamH I、XhoI、T4DNA连接酶,Takara公司;DNA 标记物、蛋白质标记时物、PCR 主混合物、ExRed核酸电泳染料、10×SDS-PAGE电泳液,北京庄盟国际生物科技有限公司;提质粒试剂盒、胶回收试剂盒,OMEGA Biotek公司;二喹啉甲酸(bicinchoninic acid,BCA)蛋白浓度测定试剂盒,碧云天生物技术公司;镍-次氮基三乙酸(nickel-nitrilotriacetic acid,Ni-NTA)Agarose,GE Healthcare;Anti-His Tag、羊抗小鼠IgG-HRP、异丙基-β-D-硫代半乳糖苷(isopropyl-β-D-thiogalactoside,IPTG)、聚偏二氟乙烯(polyvinylidene fluoride,PVDF)膜、电化学发光(electrochemiluminescence,ECL)Plus发光液,北京索莱宝科技有限公司;0.22 μm滤膜,美国Merck Millipore;其他试剂均为国产分析纯。
1.1.3 培养基
Luria-Bertani(LB)培养基用于培养重组大肠杆菌,其配制方法如下(质量分数):胰蛋白胨1%,酵母提取物0.5%,NaCl 1%,琼脂粉1.7%(固体培养基用);pH调为7.0。使用时添加卡那霉素终质量浓度至50 mg/mL,氨苄青霉素的终质量浓度为100 mg/mL。
1.2 实验方法
1.2.1 (A6K)15编码基因的设计与合成
根据目标多肽序列(A6K)15设计其编码DNA序列如下所示,在氨基酸序列之前引入6×His标签,在DNA序列首尾添加BamH I和XhoI酶切位点,同时,为便于合成,对丙氨酸A的密码子进行优化。基因合成委托北京华大基因科技有限公司进行。
5′GGATCCCATCATCATCATCATCATAGCAGCGGCCTGGTTCC-TAAAGCAGCGGCAGCAGCGGCAAAAGCAGCGGCAGCGGCA-GCAAAAGCGGCAGCAGCAGCAGCAAAAGCAGCGGCAGCGGC-AGCAAAAGCAGCGGCAGCGGCGGCAAAAGCAGCAGCAGCAG-CAGCGAAAGCAGCAGCGGCAGCAGCGAAAGCAGCAGCAGCG-GCAGCAAAAGCAGCAGCAGCAGCGGCAAAAGCGGCGGCAGC-GGCGGCAAAAGCAGCAGCAGCAGCGGCAAAAGCAGCGG-CGGCAGCGGCAAAAGCAGCAGCAGCAGCGGCAAAAGCAGCAGC-AGCGGCGGCAAAAGCAGCGGCGGCAGCAGCAAAACTCGAG-3′
1.2.2 基因的克隆及重组表达载体的构建与转化
PCR扩增(A6K)15编码基因反应条件为:95 ℃预变性3 min;95 ℃变性30 s,52 ℃退火30 s,72 ℃延伸30 s,30个循环;最后72 ℃延伸10 min。PCR产物经琼脂糖凝胶电泳验证后进行切胶回收,得到目的基因(A6K)15。
构建酶切体系,将PCR产物与空载体pET-28a(+)、pET-28a-CLCⅠ、pET-28a-SUMO、pT7-GST-His进行BamH I和XhoI双酶切,酶切产物经琼脂糖凝胶电泳验证后切胶回收,加入T4DNA ligase及缓冲液后配成连接体系,置于金属浴中16 ℃过夜连接,42 ℃热激法将连接好的重组表达质粒转化进克隆宿主E.coliDH5α感受态,在含相应抗生素的抗性平板上筛选转化子,挑选阳性单克隆进行菌落PCR鉴定。培养阳性单克隆并提取质粒,经测序鉴定正确后,再将重组质粒分别转入3种不同的表达菌株E.coliBL21(DE3)、BL21(DE3) plysS和Rosetta(DE3),菌种保藏至-80 ℃冰箱。
1.2.3 诱导表达和条件优化
将PCR鉴定及测序正确的菌株4区划线接种于LB固体培养基中,37 ℃倒置培养12~16 h,挑取单菌落接种于3 mL LB液体培养基(含相应抗性)中,37 ℃,220 r/min 振荡培养12~16 h。取培养的菌液,按1%的接种量接种于含有100 mL LB培养基(含相应抗性)的三角瓶中,37 ℃,220 r/min振荡培养至合适的OD600(0.4~0.6),用IPTG诱导蛋白表达,分别对IPTG诱导终浓度(0、0.4、0.6、0.8、1.0、2.0 mmol/L),诱导温度(15、23、30、37、40 ℃),诱导时间(3、5、7、12、24 h)进行优化,以不含(A6K)15基因的空载体为空白对照,所有样品通过15%的SDS-PAGE结果显示。
1.2.4 (A6K)15表达形式的鉴定[6]
按优化后的条件,取培养的菌液,按1%转接于200 mL液体培养基培养至OD600=0.6,加IPTG至终浓度为1.0 mmol/L,30 ℃诱导12 h。4 ℃收集菌体,取1 mL菌体离心后取沉淀和发酵上清液分别进行SDS-PAGE分析,另取100 mL菌体经PBS(20 mmol/L NaH2PO4,20 mmol/L Na2HPO4)洗涤2次后用10 mL PBS重悬,在冰水混合物中用超声细胞破碎仪进行细胞破碎(超声5 s,间隔5 s冰浴超声20 min),破碎后离心取上清液和沉淀分别进行SDS-PAGE分析其表达形式。
1.2.5 (A6K)15可溶性表达量的提高
除外源蛋白本身的性质以外,大肠杆菌的生长环境也是影响外源蛋白的可溶性表达的关键因素之一[12]。在不影响培养基pH和大肠杆菌生长的前提下,向培养基中加入磷酸盐缓冲液、微量元素(Zn、Mg等)可以提高目的蛋白的可溶性表达[12]。因此,向LB培养基中分别加入终浓度为50 mmol/L的K3PO4缓冲液、2.0 mmol/L CaCl2·2H2O,以不加磷酸盐缓冲液和微量元素的LB培养基为对照,相同条件下诱导表达(A6K)15,通过SDS-PAGE分析对比表达效果。
1.2.6 (A6K)15的纯化
由于(A6K)15中的His标签可与Ni柱中的硫酸镍结合,在蛋白上样后,带有组氨酸标签的蛋白可以特异性结合到Ni柱里,其他的杂蛋白流出。因此可采用Ni亲和层析柱(Ni-chelating column)对(A6K)15进行纯化。按优化后的条件扩大培养菌体,取45 mL培养的菌液,4 ℃、10 000 r/min离心收集菌体,用pH 7.4的PBS(20 mmol/L NaH2PO4,20 mmol/L Na2HPO4,0.9% NaCl,20 mmol/L咪唑)洗涤2次并重悬菌体,在冰水混合物中进行细胞破碎。将破碎后的细胞12 000 r/min离心10 min,收集上清液,经0.22 μm滤膜过滤。用10倍柱体积结合缓冲液(binding buffer)(20 mmol/L pH 7.4 Na3PO4缓冲液,500 mmol/L NaCl,20 mmol/L咪唑)平衡柱子,待基线平衡后再将破碎后的上清液缓慢注入系统,用洗脱缓冲液(elution buffer)(20 mmol/L pH 7.4 Na3PO4缓冲液,500 mmol/L NaCl,500 mmol/L咪唑)进行梯度洗脱(咪唑浓度分别为100、200、300、400和500 mmol/L)收集目的蛋白,流出液留样。采用SDS-PAGE分析多肽的纯化过程。
1.2.7 Western blot分析
Western blot的基本原理是通过特异性抗体对SDS-PAGE处理过的蛋白样品进行转膜着色,再通过分析抗原抗体结合的位置和信号强度来获得目的蛋白在样品中的表达情况信息[13-14]。本实验所获得的(A6K)15中含有6×His,因此选取对His特异性的一抗二抗,可检测出(A6K)15的表达情况。取10 μL蛋白洗脱收集液进行15% SDS-PAGE,将电泳所得凝胶110 V恒压冰浴电转至PVDF膜,用含有5%脱脂奶粉的TBST封闭液在脱色摇床上室温封闭2 h后,向TBST中加入鼠抗MBP一抗(1∶5 000)4 ℃孵育过夜。TBST漂洗3次,每次10 min。再加IgG-HRP标记的羊抗小鼠二抗(1∶2 000)37 ℃孵育2 h,TBST漂洗6次,每次5 min,采用ECL化学发光法显影。
1.2.8 BCA法测定(A6K)15浓度
制备蛋白标准品:取1.2 mL蛋白标准配制液加入到1管蛋白标准(30 mg牛血清蛋白)中,充分溶解配制成25 mg/mL的蛋白标准溶液,取适量此标准溶液稀释至终质量浓度为0.5 mg/mL。制备BCA工作液:V(BCA试剂A)∶V(BCA试剂B)=50∶1,充分混匀制成工作液。将标准品按0、1、2、4、8、12、16、20 μL加到96孔板的标准品孔中,加标准品稀释液补足到20 μL,纯化后的(A6K)15和稀释10倍后的全细胞蛋白各取20 μL到96孔板的样品孔中,各孔加入200 μL BCA工作液,37 ℃放置30 min。用酶标仪测定A562的吸光度,根据标准曲线和样品体积计算出(A6K)15浓度。
2 结果与分析
2.1 PCR扩增(A6K)15基因
以设计合成的pET-28a-(A6K)15作为模板,添加特异性引物进行PCR,扩增产物经1%琼脂糖凝胶电泳,在250~500 bp可见单一扩增条带,与理论(A6K)15基因片段碱基对数366 bp相符。
2.2 重组质粒的构建和酶切验证
PCR产物与空载体共同进行BamH I和XhoI的双酶切,酶切产物经琼脂糖凝胶电泳验证后分别切胶回收,将(A6K)15基因分别与质粒用连接酶进行连接,构建成为重组表达载体,转化至克隆宿主E.coliDH5α,扩大培养后提取质粒,再次用BamH I 和XhoI进行双酶切鉴定,产物用1%琼脂糖凝胶电泳验证(图1)显示,(A6K)15基因已成功克隆到载体上。

M-核酸分子质量marker;1-pT7-GST-His-(A6K)15; 2-pET28a-SUMO-(A6K)15;3-pET28a-CLCⅠ-(A6K)15;4-pET28a-(A6K)15图1 重组质粒酶切验证电泳图Fig.1 Verification of recombinant plasmids by restriction endonuclease digestion
将阳性重组质粒委托北京华大生物科技有限公司测序分析,经比对,序列正确率为100%。
2.3 诱导表达及条件优化
2.3.1 不同表达载体和宿主细胞对(A6K)15的表达影响
研究不同宿主细胞对(A6K)15的表达影响,DE3是溶源性的λDE3,所以在lacUV5启动子下携带有T7 RNA聚合酶的染色体拷贝。因此以3种不同的宿主细胞:BL21(DE3)、BL21(DE3)plysS和Rosetta(DE3)为对象[15],将含有荧光蛋白的重组融合表达载体pET-28a-CLCⅠ-(A6K)15进行转化。诱导表达后取发酵液的荧光强度进行定性定量分析,通过紫外成像仪初步观察荧光强度,已知Cerulean荧光蛋白的激发波长为433 nm,吸收波长为475 nm,通过酶标仪对发酵液进行荧光信号检测。结合图2-a荧光强度的初步观察和表1荧光信号的定量检测,结果显示,融合蛋白CLCⅠ-(A6K)15在BL21(DE3)中的表达量较其他2种宿主最高。因此选择BL21(DE3)作为表达宿主对(A6K)15进行表达。将不同的重组表达载体pET28a-(A6K)15、pET-28a-SUMO-(A6K)15、pT7-GST-His-(A6K)15、pET-28a-CLCⅠ-(A6K)15转化至BL21(DE3)进行诱导表达,SDS-PAGE结果显示,载体pET-28a(+)不能表达(A6K)15,添加融合标签(SUMO、GST、荧光蛋白CLCⅠ)的载体可以与(A6K)15形成融合蛋白,从而促进(A6K)15表达(图2-b),使用电泳胶条带定量分析软件Band Scan 5.0对各电泳泳道进行扫描,得到泳道3中融合蛋白占细菌总蛋白最高,为46.2%,由此可见,SUMO标签对促进(A6K)15的表达效果更好,因此选用pET-28a-SUMO-(A6K)15进行后续的优化实验。

a-不同菌株荧光强度;b-不同载体融合蛋白表达差异 a:从左至右依次为Rosetta(DE3)、BL21(DE3)和BL21(DE3) plysS b:M-蛋白分子质量marker;1-对照;2-pET28a-CLC-(A6K)15; 3-pET28a-SUMO-(A6K)15;4-pT7-GST-His-(A6K)15;5-pET28a-(A6K)15图2 不同宿主细胞和表达载体对融合蛋白(A6K)15的 表达差异Fig.2 Expression differences of fusion protein (A6K) 15 in different host cells and expression vectors

表1 不同宿主表达融合蛋白的荧光强度比较Table 1 Fluorescence signal detection of CLC I-(A6K)15 in different hosts
2.3.2 IPTG浓度对(A6K)15表达的影响
取培养的菌液,按1%的接种量转接于50 mL液体LB培养基中,于37 ℃扩大培养至OD600=0.6,诱导温度为37 ℃。加IPTG至终浓度分别为0、0.4、0.6、0.8、1.0和2.0 mmol/L,SDS-PAGE结果显示(图3),未添加IPTG诱导时未见有(A6K)15表达条带,在添加不同浓度IPTG后在40 kDa左右均见有不同程度的(A6K)15表达,不同终浓度IPTG对(A6K)15的表达影响不大。使用Band Scan 5.0对各电泳泳道进行扫描,得到泳道5中融合蛋白占细菌总蛋白最高,为29.8%,因此可得出,终浓度为1.0 mmol/L时(A6K)15表达量最高,可见IPTG为诱导表达的最适浓度为1.0 mmol/L。
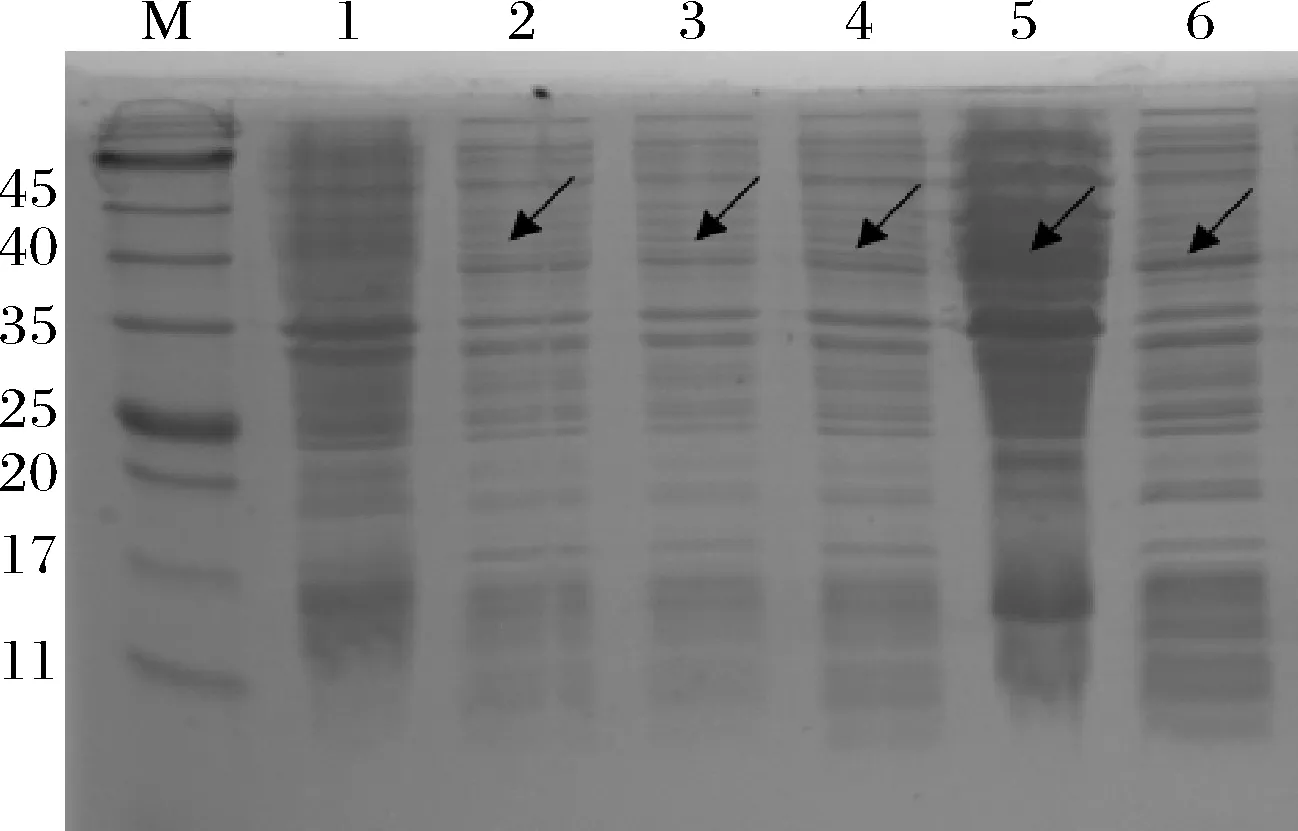
M-蛋白分子质量marker;1~6-IPTG浓度依次为 0、0.4、0.6、0.8、1.0、2.0 mmol/L图3 IPTG浓度对SUMO-(A6K)15表达的影响Fig.3 The expression of SUMO-(A6K)15 in different IPTG concentration
2.3.3 诱导温度对(A6K)15表达的影响
取培养的菌液,按体积1%转接于50 mL培养基中,于37 ℃扩大培养至OD600=0.6,加IPTG终浓度至1.0 mmol/L,分别在15、23、30、37、40 ℃条件下诱导。使用Band Scan 5.0对各电泳泳道进行扫描,可知30 ℃时目标蛋白占细菌总蛋白最高,为27.6%(图4),高温和低温都不利于(A6K)15的表达。
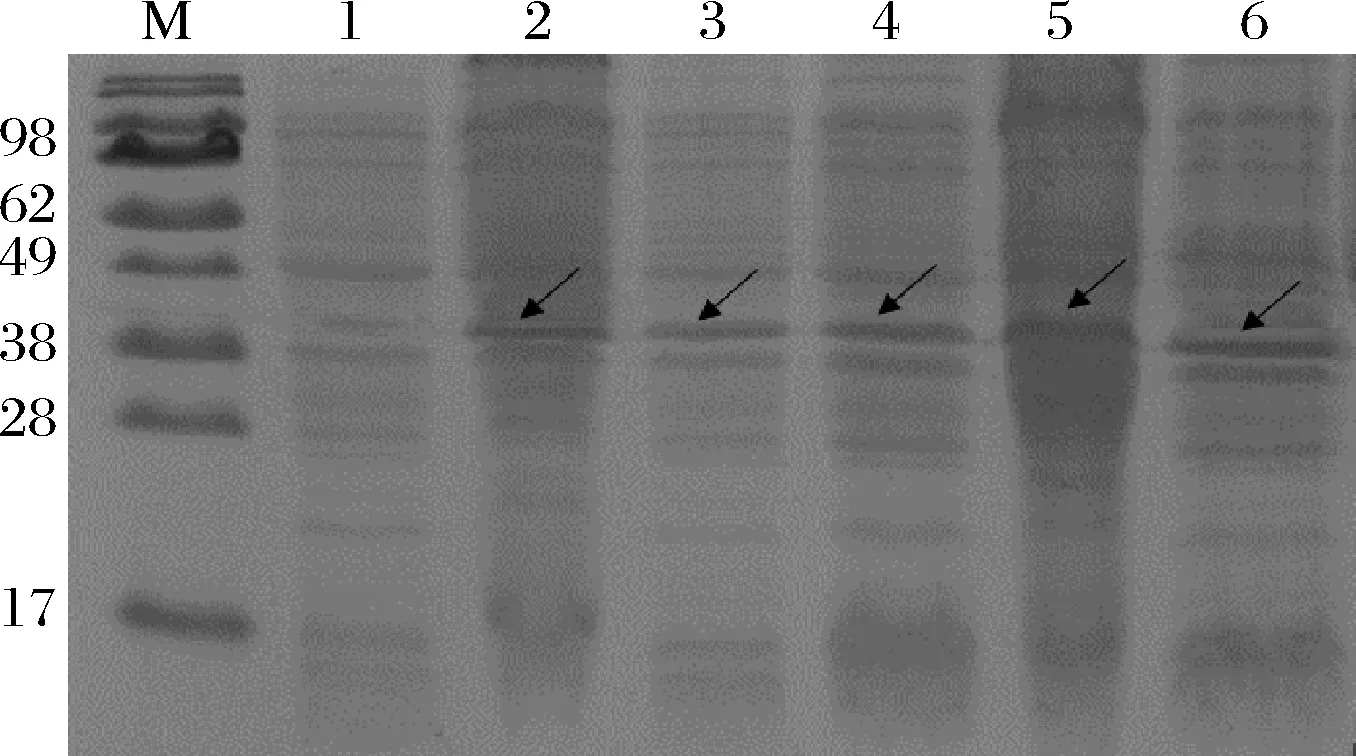
M-蛋白分子质量marker;1-空白对照;2~6-诱导温度 依次为15、23、30、37、40 ℃图4 诱导温度对SUMO-(A6K)15表达的影响Fig.4 The expression of SUMO-(A6K)15 at different inducing temperature
2.3.4 诱导时间对(A6K)15表达的影响
取培养的菌液,按体积1%转接于50 mL培养基中,于37 ℃扩大培养至OD600=0.6,添加IPTG至终浓度1.0 mmol/L,诱导温度为30 ℃,分别诱导3、5、7、12、24 h。在诱导前期,表达量随诱导时间增加而上升,12 h后趋于稳定(图5),使用电泳胶条带定量分析软件Band Scan 5.0对各电泳泳道进行扫描,可知诱导12 h时目标蛋白(A6K)15表达量最大,占细菌总蛋白的32.6%。

M-蛋白分子质量marker;1~5-诱导时间依次为 3、5、7、12、24 h;6-空白对照图5 诱导时间对SUMO-(A6K)15表达的影响Fig.5 The expression of SUMO-(A6K)15 at different inducing time
2.4 (A6K)15表达形式的定位
取经超声破碎前后的菌体离心后的上清液和沉淀进行SDS-PAGE分析,如图6所示,诱导后的全细胞蛋白中有明显的(A6K)15表达,发酵液上清液则没有特异性表达带,说明目的多肽(A6K)15为胞内表达。此外,对菌体进行超声细胞破碎后,在沉淀中可见明显的特异性条带,上清液中有微量(A6K)15表达,经Band Scan 5.0扫描分析后发现沉淀中(A6K)15含量(52.1%)显著高于上清液(20.5%),说明其主要以包涵体的形式表达。
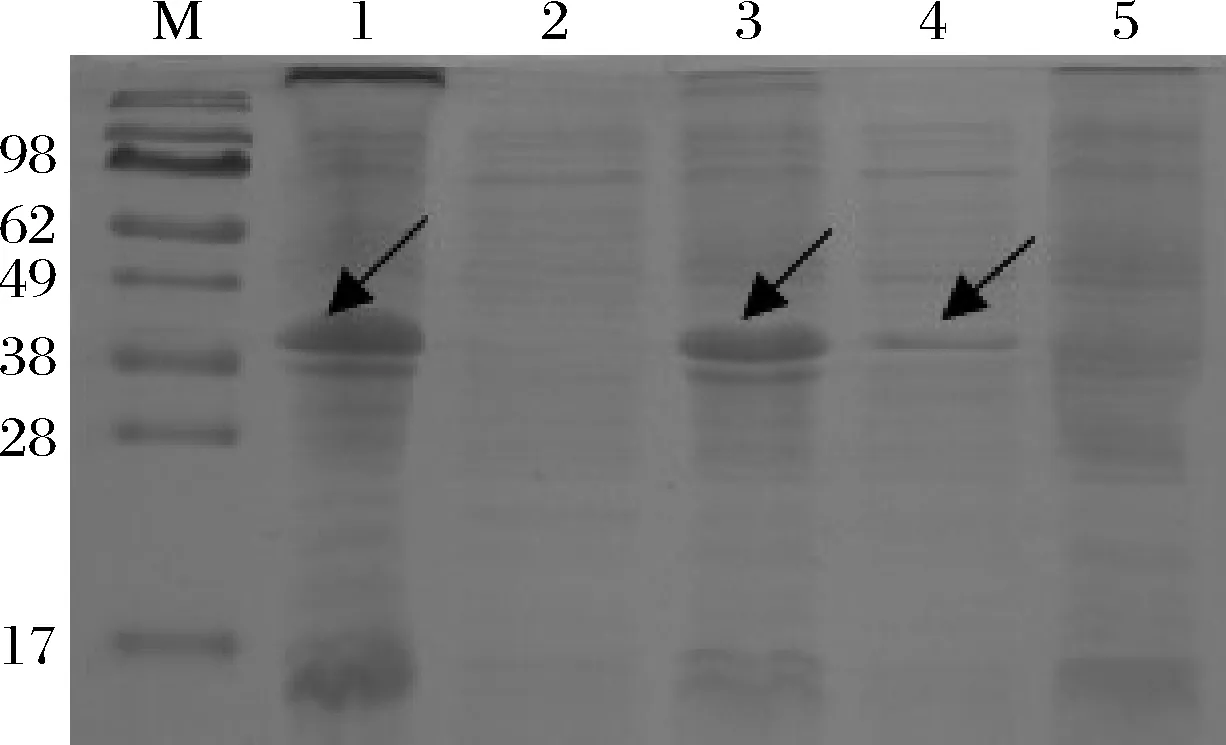
M-蛋白分子质量marker;1-诱导后菌体沉淀;2-菌体离心后上清液; 3-菌体超声破碎后沉淀;4-菌体超声破碎上清液;5-空白对照图6 表达产物在细胞中的定位Fig.6 Cell location ofSUMO-(A6K)15
2.5 (A6K)15可溶性表达量的提高
按照1.2.5所述方法对培养基进行优化,向LB培养基中加入终浓度为50 mmol/L的K2HPO4·3H2O、2.0 mmol/L CaCl2·2H2O,以未优化培养基为对照组,相同的培养条件诱导表达(A6K)15,诱导后的发酵液经超声破碎后取上清液进行SDS-PAGE分析。如图7所示,经Band Scan 5.0灰度扫描分析结果显示,培养基中分别添加K2HPO4·3H2O和CaCl2·2H2O后表达(A6K)15分别占菌体总蛋白的46.3%和65.4%,与对照组的36.8%相比,表达量有所提高。因此可证明,向培养基中添加适量的磷酸盐缓冲液和微量元素可以一定程度提高(A6K)15的可溶性表达,且添加微量元素Ca2+后表达量提高较多且杂蛋白较少。
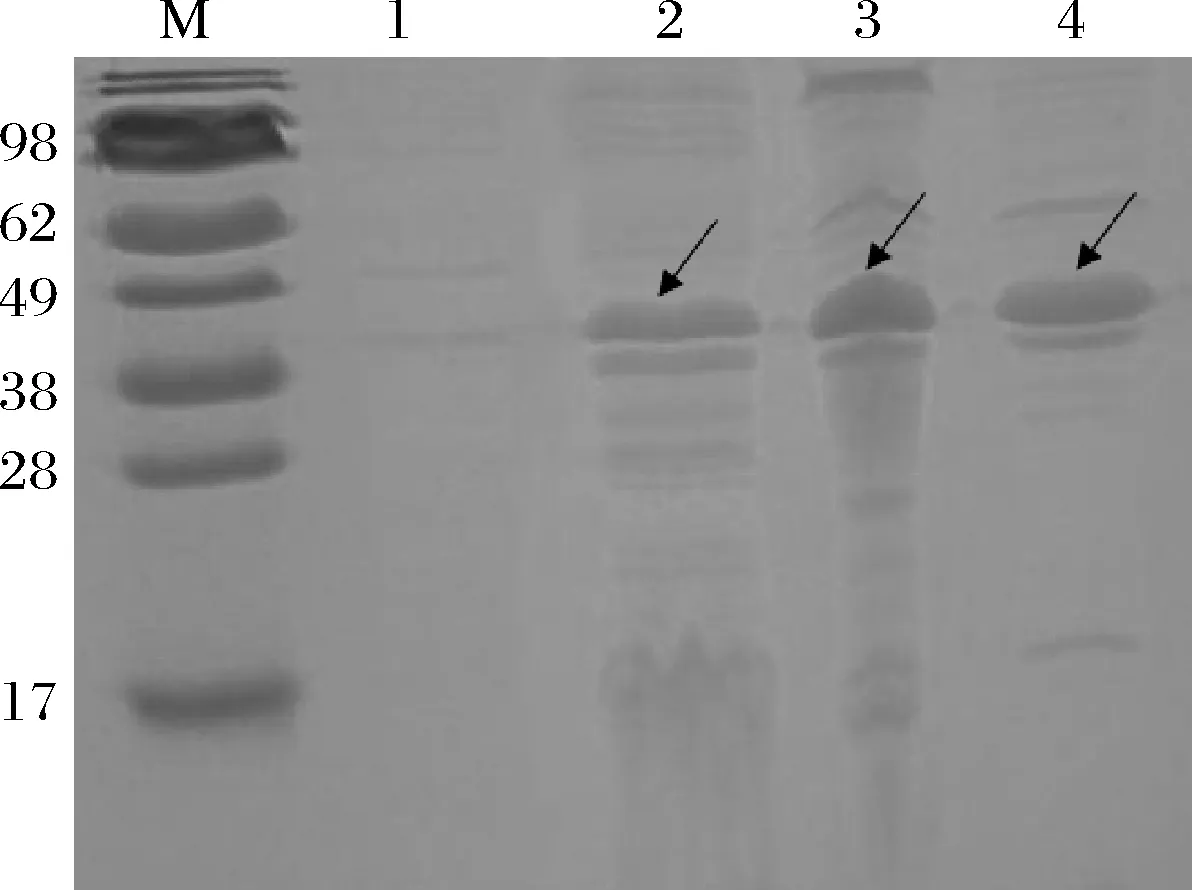
M-蛋白分子质量marker;1-空白对照;2-LB液体培养基; 3-LB添加K2HPO4·3H2O;4-LB添加CaCl2·2H2O图7 培养基组份对SUMO-(A6K)15表达的影响Fig.7 The effect of medium component on the expression of SUMO-(A6K)15
2.6 (A6K)15的纯化
按1.2.6所述方法对(A6K)15进行Ni柱亲和层析纯化。在咪唑浓度为300和500 mmol/L时有洗脱峰出现,收集出峰处的洗脱液进行SDS-PAGE分析,如图8所示,经Band Scan 5.0扫描分析结果显示,纯化前目标多肽占菌体总蛋白的37.2%,咪唑浓度为300 mmol/L时洗脱得到的目标多肽占菌体总蛋白的42.5%,咪唑浓度为500 mmol/L时目标多肽占菌体总蛋白的83.4%,因此认为咪唑浓度为500 mmol/L时洗脱得到的(A6K)15纯度较高。

M-蛋白分子质量marker;1-空白对照;2-纯化前样品; 3~4-咪唑浓度分别为300和500 mmol/L的洗脱液图8 (A6K)15纯化前后的SDS-PAGE结果Fig.8 SDS-PAGE result of (A6K)15 with purification
2.7 Western blot鉴定
纯化后的(A6K)15经1.2.7所述的Western blot分析,结果如图9所示,经Ni柱纯化后的(A6K)15具有单一的条带,说明纯化后的(A6K)15中的6×His标签能够与羊抗小鼠的多克隆抗体特异性结合,具有良好的抗原性,与预期结果一致。

1~3-Western blot 3次平行试验图9 (A6K)15的Western blot鉴定Fig.9 Western blot identification of (A6K)15
2.8 (A6K)15浓度的测定
将纯化后的蛋白通过凝胶柱脱盐除去咪唑之后,根据1.2.8方法用BCA蛋白浓度测定试剂盒测定(A6K)15的浓度。标准曲线为y=1.392 5x+0.042,R2=0.999 8,取可溶性表达的(A6K)15蛋白溶液进行吸光度测定,代入标曲计算得可溶性(A6K)15的质量浓度为0.63 mg/mL。取稀释后的全细胞蛋白溶液进行吸光度测定,计算可得蛋白浓度为5.97 mg/mL,由图6可知,(A6K)15形成的包涵体蛋白占全细胞蛋白的52.1%,由此可以计算出以包涵体形式表达的(A6K)15质量浓度高达3.11 mg/mL。因此可推算,大肠杆菌高密度发酵可生产重组蛋白(A6K)15的收率为3.7 g/L,所需试剂及耗材价格约为700元/mg,但化学合成多肽的价格则达1 000元/mg。因此,与化学合成方法相比较,生物法合成更能节约成本,且当发酵技术进一步成熟和产量达到一定规模后,若需要大规模生产,相比于化学合成法生产成本可望进一步降低。
3 结论
原核表达系统具有操作方便、快捷,耗时短,表达量大,适合工业化生产等优点,是蛋白表达和生物合成最常用的方法[16]。其中,大肠杆菌原核表达系统发展最为迅速、成熟,因为其具有操作方便、遗传背景明确、表达量高且试验成本低等优点,成为目前研究较为深入并最常用的表达系统之一[17-18]。因此本研究选用大肠杆菌作为宿主菌表达新型小分子短肽。但使用大肠杆菌表达外源蛋白质时,虽然表达量大但可溶性较低,目的蛋白常以包涵体形式存在[19],故本研究试图通过丰富培养基配方提高目的蛋白可溶性表达。
本研究选用了4种原核表达载体pET-28a(+)、pET-28a-CLCⅠ、pET-28a-SUMO、pT7-GST-His插入目的基因,将构建好的重组表达载体进行诱导表达后发现,不带有融合标签的pET-28a(+)无法表达(A6K)15,带有融合标签的载体均可不同程度地与(A6K)15形成融合蛋白,从而促使目标多肽进行表达。其中SUMO标签对(A6K)15的表达具有最为显著的促进效果,SUMO作为一种新型的融合载体,它能提高目的蛋白的折叠程度和溶解度[20-21]。并且SUMO蛋白酶能够特异性识别SUMO的三级结构,这样就能够避免对目的多肽的切割,这对于后续分离纯化时保持目标多肽的活性至关重要[22-23]。因此,以SUMO为融合标签形成融合蛋白的方式表达,不仅可避免(A6K)15对宿主的杀伤作用,也保护了(A6K)15免受蛋白酶的降解。综合以上原因,选择pET-28a-SUMO作为最优表达载体。与此同时,通过荧光蛋白与(A6K)15结合后在不同表达宿主上进行荧光信号检测,得到其最优表达宿主为BL21(DE3)。在此基础上,利用大肠杆菌表达系统对(A6K)15进行诱导表达,为探索此新型小分子短肽的最优表达条件,进一步通过设置不同终浓度IPTG以及不同诱导温度和时间诱导(A6K)15,筛选出(A6K)15在E.coliBL21(DE3)中表达量最高的条件是:添加终浓度为1.0 mmol/L的IPTG、30 ℃条件下诱导12 h。此外,本研究还在传统液体LB培养基的基础上添加微量元素和磷酸盐缓冲液,在一定程度上有效提高了(A6K)15的可溶性表达。
本研究虽然解决了新型表面活性肽(A6K)15在大肠杆菌中异源表达的关键问题,但对此类表面活性肽的研究还只是一个开始。虽然对提高(A6K)15的可溶性表达进行了初步探索,但是SDS-PAGE结果显示,(A6K)15在表达时仍主要以包涵体形式存在,因此未来需要解决(A6K)15在表达中出现的包涵体问题,以提高表达量、浓度和纯度。用SUMO蛋白酶特异性切割去除SUMO标签,继而用胰蛋白酶对(A6K)15重复序列进行切割以获得单一氨基酸序列的小肽A6K,旨在获得纯度较高的具有利用价值的表面活性肽,并且与化学合成相比进一步降低生产成本,为生物发酵法合成表面活性肽提供成熟的研究思路和技术路线。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:38
黑龙江大学自然科学学报(2021年4期)2021-11-19 07:05:02
石油沥青(2021年4期)2021-10-14 08:50:52
现代园艺(2017年13期)2018-01-19 02:28:09
中国调味品(2017年2期)2017-03-20 16:18:21
现代检验医学杂志(2016年3期)2016-11-15 01:59:48
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
合成化学(2015年9期)2016-01-17 08:57:24
合成化学(2015年10期)2016-01-17 08:56:33
药学与临床研究(2015年4期)2015-06-05 11:35:54
