
组蛋白乙酰化修饰在哮喘发病机制中的研究进展
2022-02-17 04:32:24陈绍鹏曾敏娟赖天文
中国全科医学 2022年6期
陈绍鹏,曾敏娟,赖天文*
哮喘是一种慢性呼吸系统疾病,包括异常的气道炎症、气道重塑和气道高反应性(AHR),近20年来其发病率从5.5%上升至约8%,给患者家庭及社会带来极大的经济负担[1]。组蛋白乙酰化是目前研究最为广泛和深入的表观遗传修饰之一,主要指组蛋白乙酰转移酶(histone acetyltransferase,HAT)和组蛋白去乙酰化酶(histone deacetylase,HDAC)的调节作用,调节失衡会引起基因调控紊乱,导致慢性炎症[2]、自身免疫性疾病[3]、癌症[4]等疾病的发生。研究表明HAT、HDAC的改变与哮喘密切相关[5-6]。本文对近年来组蛋白乙酰化修饰在哮喘发病机制中的研究作一综述,为临床治疗提供依据。
1 组蛋白乙酰化概述
组蛋白乙酰化是指在组蛋白氨基端尾部的赖氨酸ε位的氨基处加上乙酰基,该过程可逆。
HAT是将乙酰辅酶A(CoA)的乙酰基转移至组蛋白N端尾部赖氨酸残基上的一组酶。当HAT转移乙酰基到组蛋白的赖氨酸残基上时,带负电荷的乙酰基打破原来组蛋白的平衡,导致脱氧核糖核酸(DNA)序列结构松弛和展开,促进转录因子与DNA序列的接触,使DNA序列的转录增强[7]。哺乳动物主要有3个HAT家族:p300/CREB结合蛋白(p300/CBP)家族、MYST家族和GNAT家族。
HDAC是去除赖氨酸残基上乙酰基的一类蛋白酶。当HDAC去除组蛋白赖氨酸残基中的乙酰基后,组蛋白恢复原来的正电荷,与带负电荷的DNA紧密结合,使转录因子难以接近启动子,从而抑制特定基因的转录。HDAC分为四类:Ⅰ类(HDAC 1-3和8)、Ⅱ类(HDAC 4-7、9和10)、Ⅲ类(SIRT1-7)和IV类(HDAC11)[8]。Ⅰ、Ⅱ和Ⅳ类为锌离子(Zn2+)依赖型HDAC,具有相似的去乙酰基结构域,表明单个化合物可以同时抑制Zn2+依赖型HDAC。而Ⅲ类HDAC在催化过程中依赖于烟酰胺腺嘌呤二核苷酸(nicotinamide adenine dinucleotide,NAD+)。
2 组蛋白乙酰化与哮喘
2.1 HAT在哮喘发病中的作用 p300促进/加重哮喘发病,HAT与哮喘密切相关。目前关于HAT对哮喘发病机制的研究主要集中在p300。p300介导ORMDL3组蛋白乙酰化,加重哮喘气道炎症和气道重塑。p300选择性抑制剂——C646抑制哮喘小鼠p300的表达,ORMDL3的表达降低,从而缓解AHR和气道重塑[5]。p300对核因子活化B细胞κ轻链增强子(NF-κB)p65的乙酰化使β-连环蛋白(β-catenin)促进p65的核转位,进而促进了人气道平滑肌(ASM)细胞中白介素(IL)-6的表达[9]。p300依赖的组蛋白H3乙酰化参与了凝血酶诱导的人肺上皮细胞IL-8的表达[10]。细颗粒物暴露和冷应激(PMCS)联合诱导使p300表达显著上调,进而增加CD4+T淋巴细胞IL-4基因启动子中的H3K9和H3K14乙酰化水平,加重哮喘小鼠的炎性反应和氧化还原反应[3]。
p300在哮喘模型大鼠肺组织分离培养出的CD4+T淋巴细胞中的表达增加,Notch1启动子组蛋白的乙酰化水平升高,IL-4、IL-5、IL-13以及NF-κB和AP-1水平亦显著升高,可见p300通过Notch1启动子组蛋白的乙酰化影响哮喘大鼠肺CD4+T淋巴细胞的分化[11]。p300催化H3K27的乙酰化,促进IL-9的强烈表达,诱导辅助T淋巴细胞9(Th9细胞)分化,并介导过敏性气道炎症[12]。此外,P300和CBP调节β-Catenin信号有利于黏液细胞的分化[13]。综上所述,p300可介导Notch1、β-Catenin基因及H3K27位点的乙酰化,影响Th细胞及黏液细胞的诱导分化。
2.2 HDAC 在哮喘发病中的作用 相比于HAT,研究者将更多的目光放在HDAC与哮喘发病的研究上。各个HDAC在哮喘发病中的表达及作用并不相同,同一种HDAC也可能起着不同甚至相反的作用。
2.2.1 Zn2+依赖型HDAC
2.2.1.1 HDAC5、HDAC6、HDAC8促 进 /加 重 哮 喘 发 病
在卵清蛋白(OVA)诱导的过敏性哮喘小鼠肺组织中,HDAC5、HDAC6、HDAC8的表达水平明显升高,支气管肺泡灌洗液(BALF)中IL-4和IL-5水平升高[14]。HDAC8抑制剂PCI可以同步降低HDAC8和半乳糖凝集素-3(Gal-3)的表达、减少M2巨噬细胞极化,改善AHR和过敏性气道炎症[15]。
2.2.1.2 HDAC2、HDAC3、HDAC7、HDAC10、HDAC11抑制/减轻哮喘发病 哮喘患者外周血单个核细胞HDAC2的信使核糖核酸(mRNA)表达水平在重度哮喘患者中明显低于非重度哮喘患者[16]。室尘螨(HDM)诱导的HDAC2+/-小鼠炎性细胞浸润显著增强、肺组织中T辅助因子和IL-17A表达水平升高。IL-17A缺失或抗IL-17A治疗逆转了由HDAC2损伤引起的气道炎症增强[17]。
萝卜硫素通过上调核因子E2相关因子2(Nrf2)的表达和增强HDAC2的活性,恢复了类固醇的敏感性[18]。在重度哮喘患者中,HDAC2可能通过介导类固醇来关闭活化的炎症基因,且磷酸肌醇3-激酶δ(PI3Kδ)的活化使得HDAC2的活性和表达被氧化应激降低,因此可通过PI3Kδ抑制剂增加HDAC2的表达从而逆转类固醇耐药[19]。
在16-HBE细胞中,香烟烟雾提取物(CSE)降低了HDAC2的活性、HDAC3的表达及活性、糖皮质激素受体(GR)的核转位,增加了NF-κB的核表达、1/2磷酸化胞外信号调节激酶(pERK 1/2)与1/2总胞外信号调节激酶(tERK1/2)比值以及炎性因子mRNA的表达,加重了炎性反应[20]。
低表达HDAC7、HDAC9和HDAC10诱导的组蛋白高乙酰化状态加重嗜酸性气道炎症。糖皮质激素(GC)上调HDAC7、HDAC9、HDAC10、HDAC11,特别是HDAC10,是改善气道炎症的机制之一[21]。
2.2.1.3 HDAC1、HDAC4、HDAC9在哮喘中的作用尚未明确
在PMCS联合暴露诱导小鼠哮喘模型研究中,CD4+T淋巴细胞HDAC1明显降低[3],HDAC1在哮喘模型大鼠肺组织分离培养出的CD4+T淋巴细胞中的表达下降[11]。相反,SU等[14]报道HDAC1的表达水平在OVA诱导的过敏性哮喘小鼠肺组织中明显升高,哮喘小鼠气道阻力显著增强,BALF中IL-4和IL-5水平升高。
转化生长因子-β1(TGF-β1)通过降低微小核糖核酸-206(miR-206)水平,上调HDAC4表达,进而上调周期蛋白D1(cyclin D1)表达,诱导气道平滑肌细胞(ASMCs)增殖,加重气道重塑[22]。在佛波酯(PMA/A23187)诱导人肥大细胞(HMC-1)发生过敏性炎症中,HDAC4作为miR-20a的靶基因表达升高,促进肿瘤坏死因子(TNF)-α、IL-1β等的分泌[23]。相反,SU等[14]报道HDAC4的表达水平在OVA诱导的过敏性哮喘小鼠肺组织中显著下降,肺组织中可见炎性细胞浸润、黏液堆积,BALF中IL-4和IL-5水平升高。
哮喘患者中HDAC9 mRNA表达水平与疾病严重程度呈正相关[24]。相反,ZHANG等[21]报道低表达HDAC9诱导的组蛋白高乙酰化状态可加重嗜酸性气道炎症。
2.2.2 NAD+ 依赖型HDAC
2.2.2.1 SIRT2、SIRT7促进/加重哮喘发病 哮喘患者肺组织中SIRT2表达升高,发挥促炎作用,加重哮喘症状[25]。SIRT2促进了三种过敏原〔尘螨、豚草、烟曲霉(DRA)〕诱导嗜酸粒细胞的招募[26]。此外,SIRT2抑制剂可减少巨噬细胞的交替激活,并显著消除三种过敏原诱导的肺部炎症[1]。SIRT7通过调节转化生长因子-β受体Ⅰ(TGFβR1)的表达,参与TGF-β1诱导的ASM细胞增殖和迁移,提示SIRT7加重哮喘气道重塑[27]。
2.2.2.2 SIRT6抑制/减轻哮喘发病 SIRT6的抗炎作用与T淋巴细胞中转录因子GATA3乙酰化水平降低及Th2免疫应答的减少有关[25]。过表达SIRT6可显著减弱OVA诱导的浸润肺组织中的炎性细胞,细支气管黏液积聚可被有效降低至接近正常水平[28]。此外,SIRT6的过表达影响TGF-β1诱导的上皮细胞-间充质转化(EMT)标志物变化和EMT样细胞行为,降低了细胞的迁移和增殖速率,改善了气道重塑[29]。
2.2.2.3 SIRT1抑制哮喘炎性反应 在人支气管上皮细胞(16HBE细胞)及OVA诱导小鼠的哮喘模型中SIRT1的mRNA和蛋白水平均下调,且SIRT1的下调使IL-6的表达升高,而这一表达可被蛋白激酶B(Akt)抑制剂及SIRT1激活剂逆转[6,30]。此外,SIRT1作为miR-221的靶基因,可逆转哮喘患者HBE细胞损伤[31]。SIRT1抑制了环境颗粒物(PM)暴露后的胆固醇调节元件结合蛋白1(SREBP1),并进一步下调了铁结合核蛋白(PIR)和Nod样受体蛋白3(NLRP3)炎性小体,减轻人肺成纤维细胞炎症[32]。
3 HDAC抑制剂或激活剂治疗哮喘的研究进展
3.1 HDAC抑制剂治疗哮喘 已有研究报道,HDAC6抑制剂——曲古霉素A(TSA)抑制HDAC,可下调适应性过敏免疫反应,减轻气道炎症。TSA减少了大气道血管周围嗜酸粒细胞数量和黏液产生,减轻了哮喘表征[33-34]。
选择性HDAC8抑制剂PCI-34051和地塞米松可减轻哮喘模型鼠的嗜酸性炎症、AHR及气道重塑[34]。
丁酸通过抑制HDAC活性来抑制固有淋巴样2型细胞(ILC2)的增殖,减少IL-13、IL-15的产生,进而改善AHR和气道炎症[35]。
研究者CAO等[36]合成了用于降解I类HDACs的蛋白水解靶向嵌合体(PROTACs),其中一种PROTACs HD-TAC7对HDAC3具有良好的降解效果,提示PROTACs具有治疗潜力。
3.2 HDAC激活剂治疗哮喘 小剂量大环内酯类药物如阿奇霉素和红霉素能恢复HDAC2的活性。OVA和香烟烟雾诱导的小鼠哮喘模型中HDAC2的蛋白水平降低,但是罗红霉素可以提高HDAC2的表达水平,以降低香烟烟雾暴露的哮喘小鼠的气道炎症[2]。
用miR-21特异性拮抗剂(Ant-21)或pan-PI3K抑制剂LY294002治疗,可以降低PI3K活性并恢复HDAC2水平,进而抑制AHR和恢复类固醇对过敏性气道疾病的敏感性[37]。此外,PI3K抑制剂(BZE235和LY294002)可通过恢复HDAC2活性和抑制核信号转录因子磷酸化来改善严重哮喘患者的GC不敏感性[38]。
体内外实验发现,低剂量茶碱增强了上皮细胞和巨噬细胞中HDAC的活性,浓度在1~10 μM之间的茶碱和恩丙茶碱增加了HDAC2的活性,而高于该浓度则抑制了HDAC2的活性,表明低剂量茶碱可通过增加HDAC2的激活而发挥抗哮喘作用[39]。
穿心莲内酯通过恢复HDAC活性,提高HDAC2蛋白质水平,降低脂多糖/γ-干扰素(LPS/IFN-γ)诱导的IL-27水平和AHR,恢复地塞米松敏感性[40]。
骨化三醇能增强IL-13诱导的哮喘患者ASM细胞HDAC的表达及活性,减弱TNF-α对组胺反应的增强作用[41]。
4 小结
近年来组蛋白乙酰化修饰在哮喘发病机制中的研究概述如图1。目前,对于其在哮喘发病机制中的研究并不深入,不同研究间甚至存在矛盾之处。造成此种现象的原因可能与HAT和HDAC基因表达存在动态变化,或是不同的研究使用的体内外模型存在差异。另一方面,HDAC抑制剂或激活剂能调控哮喘的炎性反应,为临床靶向治疗哮喘提供了理论依据。
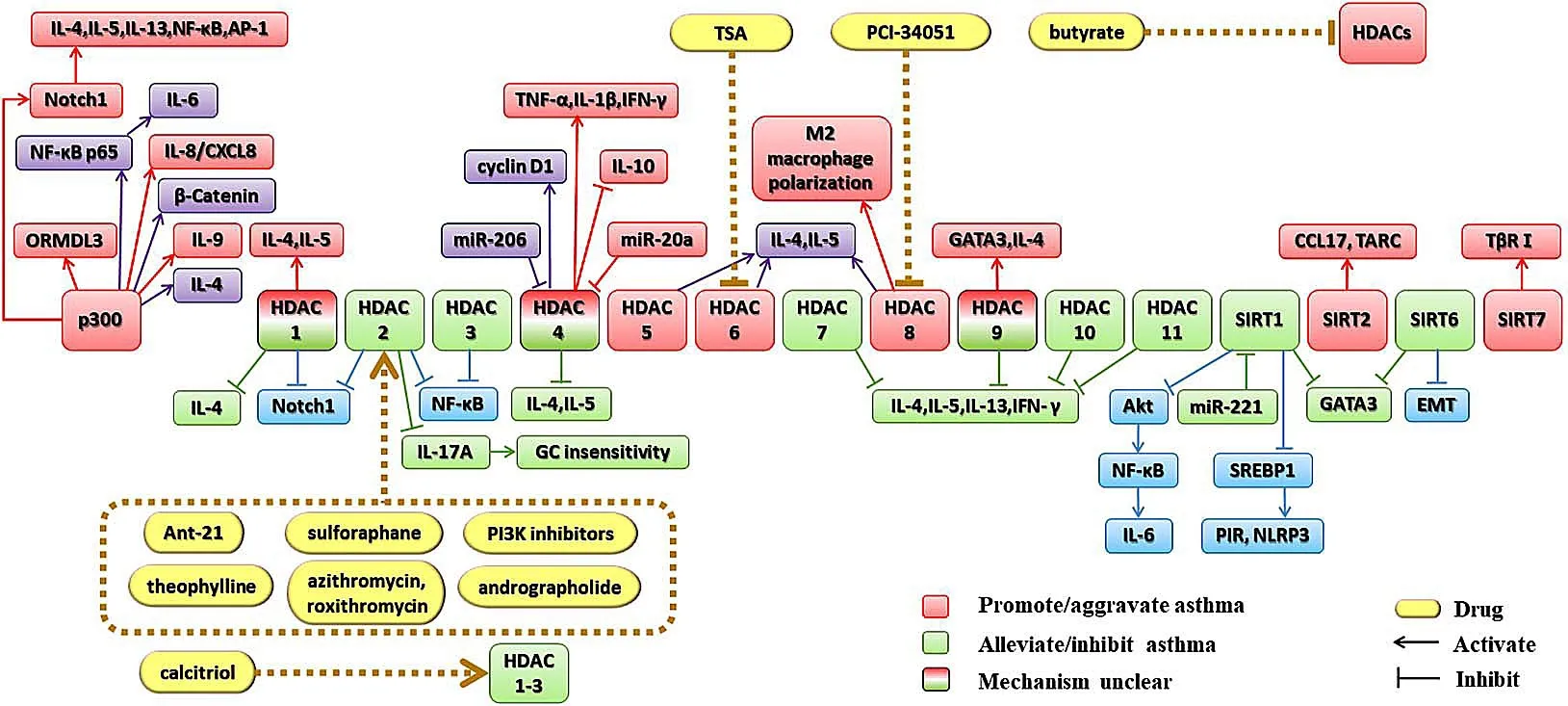
图1 组蛋白乙酰化修饰在哮喘发病机制中的研究进展概况Figure 1 Recent five-year advances in histone acetylation in the pathogenesis of asthma
作者贡献:陈绍鹏进行检索文献、撰写论文并对文章负责;曾敏娟进行排版、作图;赖天文进行质量控制及审校。
本文无利益冲突。
本文文献检索策略:
使用PubMed网站的查询框在“All Fields”范围内以“(HAT) AND (asthma)”“(p300) AND (asthma)”“(HDAC)AND (asthma)”和“(SIRT) AND (asthma)”为关键词检索文献,重点关注近5年发表的文献。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08 00:39:50
国际呼吸杂志(2019年22期)2019-12-09 09:20:26
国际呼吸杂志(2019年5期)2019-03-30 01:38:20
国际呼吸杂志(2019年3期)2019-03-01 05:39:06
国际呼吸杂志(2019年2期)2019-02-14 06:11:26
上海农业学报(2017年3期)2017-04-10 12:39:26
中国组织化学与细胞化学杂志(2016年4期)2016-02-27 11:15:53
中国当代医药(2015年16期)2015-03-01 02:03:13
中国药理学通报(2014年2期)2014-05-09 08:22:39
中国神经精神疾病杂志(2013年1期)2013-03-11 20:23:32
