
模型炭催化加氢气化反应特性研究
2022-02-17 09:39王沁汾张建树毕继诚
燃料化学学报 2022年1期
曲 旋 ,王沁汾 ,严 帅 ,冯 俊,4 ,张建树,* ,张 荣,* ,毕继诚
(1. 中国科学院山西煤炭化学研究所 煤转化国家重点实验室,山西 太原 030001;2. 石河子大学化学化工学院 新疆兵团化工绿色过程重点实验室-省部共建国家重点实验室培育基地,新疆 石河子 832003;3. 华东理工大学 资源与环境工程学院,上海 200237;4. 中国科学院大学,北京 100049)
近年来,天然气的需求呈快速增长的趋势,开发煤制天然气技术是弥补中国天然气资源短缺的有效手段。现有的煤制天然气技术包括蒸汽-氧气两步法、催化气化和加氢气化三种[1,2],其中,以鲁奇炉为代表的蒸汽-氧气两步法和加压流化床煤催化气化技术均实现了工业化运行,而加氢气化技术仍处于中试放大阶段。从制天然气的角度来看,煤加氢气化技术路径最短,产物组分相对单一,具有较高的热效率(79.6%)和甲烷收率(40%−50%)。但是该过程需在较高的反应温度和氢压(900−1100 ℃,5−7 MPa)下进行,且过程碳转化率偏低,仅为50%−60%,这主要是由于煤结构中存在一定量缩合程度较高的芳环结构,此部分碳与氢气的反应活性较差,反应速率低[3]。
通过引入催化剂可有效解决加氢气化过程中碳的低反应性问题[4,5],同时可缓解反应所需的苛刻环境,在温和条件下(750−900 ℃,1−3 MPa)实现高的碳转化率和甲烷收率[6,7]。煤CHG 的本质为C-H2的催化反应过程,众多研究者通过对元素周期表中的元素进行探索后,发现只有碱金属(如K、Na、Li)、碱土金属(Ca)和过渡金属(如Fe、Co、Ni)元素对应的化合物能对碳-氢反应起到催化作用,并提出了相应催化作用机理[8−10]。Liu 等[11]以10%的Na2CO3、K2CO3、CaO 和CaCO3为催化剂,在固定床中于850 ℃,5.0 MPa 下研究了褐煤半焦的催化加氢气化。发现碱金属催化剂活性高于碱土金属,Na、K 等在反应过程中能够扩散至半焦的体相结构中,促进大分子缩合芳环结构的断键生成小分子芳环结构的同时抑制了炭结构的石墨化进程,使半焦中碳具有高的加氢反应性。Matsumoto等[12]研究了过渡金属催化剂对Yallourn 半焦的催化加氢气化活性,发现过渡金属催化剂Fe、Co 和Ni 的负载量在达到0.5%以上即可表现出良好的催化活性,且催化活性顺序为Co > Ni > Fe。过渡金属元素一方面通过对H2产生解离吸附的作用,提供气化反应所需的活性氢;另一方面可通过与半焦结构中的碳发生相互作用,削弱C−C 键能,从而促进了C-H2反应的进行[13]。Jiang 等[8]发现,2%CaO 的加入对烟煤半焦的催化加氢气化具有极好的催化效果,CaO在反应过程中与半焦中的碳发生结合,使碳的电子结合能发生偏移,形成新的活性较高的炭物种作为活性位进行加氢气化反应。
已有的文献研究表明,碱金属、碱土金属和过渡金属均适用于炭CHG 过程,但是关于此三大类催化剂对炭CHG 活性顺序的说法尚不统一。Hong等[14]报道在褐煤半焦的CHG 过程中,三类催化剂活性顺序为碱金属 > 过渡金属 > 碱土金属。Jiang等[15]报道在烟煤半焦CHG 过程中,三类催化剂活性顺序为碱土金属 > 碱金属 > 过渡金属。由于文献中同一种类催化剂的母体和负载方式基本相同,而催化剂显示的催化活性顺序的差异可能与原料的物理化学性质及催化剂的负载量不同有关。因此,针对C-H2反应中催化剂的活性,有必要在灰含量和硫含量极低的模型炭中负载相同量催化剂的条件下进一步进行考察。此外,炭CHG的反应条件如反应温度、氢气压力、模型炭比表面积、催化剂添加量等因素对催化剂催化活性均具有影响,有关该方面的研究仍不全面。战书鹏等[16]用褐煤考察了温度和压力对碱金属钾CHG 活性影响,发现从700−850 ℃,0.1−4.0 MPa 升高温度和氢气压力均可提高催化剂的CHG 活性;Jiang 等[8]考察了反应温度和氢气压力对碱土金属Ca 的CHG活性的影响,发现从700−800 ℃,0.1−2.25 MPa 升高温度和氢气压力,甲烷生成速率均有所提高。关于过渡金属、温度和压力对其炭CHG 的影响规律是否与碱金属和碱土金属类似仍需进一步探索。张峰等[17]研究了Fe 催化剂在不同比表面积的煤焦中分散性对炭CHG 的影响,发现增大煤焦的比表面积可促进金属 Fe 催化剂在煤焦表面的分散,金属 Fe 的平均粒径也相对减小,有利于金属Fe的还原及中间相产物 Fe3C 的生成,从而发挥催化加氢的作用,提高煤焦CHG 活性。Matsumoto 等[12]考察了Fe/Co/Ni 的负载量对褐煤半焦CHG 活性的影响,发现当负载量高于1%时,活性增加幅度减小;Haga 等[18]发现石油焦的CHG 活性随着Ni/Fe负载量从0−6%增大而显著提高。通过文献分析发现,关于催化剂负载量与炭CHG 活性的关系,可能与含碳原料的比表面积和炭结构等存在潜在的关联,因此,有必要在具备不同物理化学结构的含碳原料中进一步明确催化剂的添加量对炭CHG活性的影响。
本研究首先以活性炭为原料,在加压热天平中考察了碱金属、碱土金属、过渡金属催化剂对模型炭CHG 的催化活性。借助表征分析手段和文献中已有研究结果,初步探讨了催化剂活性差异的原因。在催化活性较优的Co 催化剂存在下,进一步探索了Co 对碳-氢反应催化活性与催化剂添加量、反应温度、氢气压力、含碳原料物理化学结构等因素的关联。借助GC、BET 和TPR-MS 等表征分析了Co 对碳-氢反应的催化作用过程。以期为煤/半焦催化加氢气化的实际应用过程中催化剂和工艺条件的选择提供理论依据和数据支撑。
1 实验部分
1.1 焦样的制备和催化剂负载
实验用沥青基活性炭(无灰、低硫、低氧)、府谷烟煤和阳泉无烟煤首先进行破碎、筛分处理,取80−120 目样品。将府谷烟煤和阳泉无烟煤采用HCl-HF 进行脱灰处理,以消除矿物质对实验的影响。具体脱灰步骤:在室温条件下称取40 g 的煤样,之后加入560 mL HCl (6 mol/L),60 ℃恒温水浴12 h(每隔0.5 h 搅拌约2 min),之后抽滤,用AgNO3溶液检验无白色沉淀生成。将获得的样品加入300 mL HF(23 mol/L) 进行洗涤与过滤,HF 洗涤和过滤等操作过程与HCl 的处理过程保持一致。脱灰后的样品于105 ℃下真空干燥12 h,备用。
制焦过程:将活性炭/脱灰府谷烟煤/脱灰阳泉无烟煤置于固定床中,于900 ℃,常压Ar 气氛下,以10 ℃/min 的升温速率升至900 ℃后,并恒温3 h。将获得的焦样分别命名为:HXT、FG 和YQ。另将获得的FG 半焦进行扩孔处理,具体步骤为使用100 mL/min 的CO2在固定床中于900 ℃进行预处理2.0 h,处理后的半焦命名为FGKK。不同样品的工业分析和元素分析见表1。
以Co(NO3)2·6H2O、 Ni(NO3)2·6H2O、 Fe(NO3)3·9H2O、Ca(NO3)3·4H2O、Mg(NO3)2·4H2O 和K2CO3作为催化剂前驱体,采用等体积浸渍法将不同比例的催化剂负载至表1 中的含碳原料上。操作过程如下:首先将称取的一定量的催化剂母体用1.0 mL蒸馏水进行溶解,取1.0 g 样品倒入催化剂溶液中,并用玻璃棒于50 ℃的超声条件下进行搅拌1.0 h,然后将获得的样品于105 ℃进行真空干燥12 h,得到负载催化剂的样品备用。金属催化剂的负载量以金属元素在半焦中的质量分数计。
1.2 催化加氢气化实验
模型炭催化加氢气化实验在加压热天平中进行,装置示意图如图1 所示,整个实验系统主要由天平头、坩埚、电加热炉和自动控制系统组成。反应温度由K 型热电偶进行控制,系统压力由背压阀控制,坩埚中样品的失重过程通过天平头进行实时测量。
首先称取20.0 mg 样品置于坩埚中,用氢气(500 mL/min)置换反应系统中的空气30 min,之后用氢气将反应系统冲压至设定压力(0.1−3.0 MPa),之后保持氢气流量为200 mL/min。待反应系统压力达到稳定后,打开电加热炉电源,以50 ℃/min的升温速率,将反应区的温度由室温升至设定温度(750−950 ℃),并于设定温度下等温反应1.0−3.0 h。为了消除浮力的影响,每个条件下的实验均进行20.0 mg 石英砂的空白实验,将每个反应条件下获取的催化加氢气化失重数据减去相应空白实验的失重数据,得到实际原料催化加氢气化的失重曲线。通过失重曲线计算样品的转化率和失重速率。

式中,m0为样品的初始质量,mt为t时刻样品的质量,m为初始样品中去除催化剂后的质量。
失重速率曲线是通过对失重曲线求导得到的,即为dx/dt。
1.3 产物分析及样品表征
活性炭、半焦中的C、H、N、S 含量通过德国的Vario EL 元素分析仪进行测定。模型炭的工业分析采用GB/T 212—2008 的方法进行。
部分加压热天平中CHG 实验的含碳产物气体(CO、CO2和CH4)通过气袋进行收集,采用离线色谱(GC-950,TCD 检测器;GC-1790, FID 检测器)进行定量分析。
在常压程序升温还原(AP-TPR,由TP5090 催化平台+质谱仪组成)装置上,于氢气气氛中进行样品的程序升温反应过程中气体产物(CO、CO2、CH4)的实时定性检测。每次样品量为100 mg,升温速率为25 ℃/min,气体流量为200 mL/min,待质谱稳定后TP5090 开始加热,TP5090 和质谱同时记录。质谱型号为 Pfeiffer Vacuum OmniStarTM/ThermoStarTMGSD 320,TP5090 催化平台由天津先权公司生产。
HXT、FG、FGKK 的比表面积和孔容使用美国麦克公司生产的 Tristar 3000 Micromeritics instrument物理吸附仪进行测定。通过对氮气吸附等温线的分析获得样品的比表面积和孔容。
2 结果与讨论
2.1 不同催化剂的催化加氢气化活性
图2 显示了在850 ℃和3.0 MPa 的氢气压力下,2%负载量的碱金属、碱土金属和过渡金属催化剂对HXT 催化加氢气化的活性。图2(a)为样品转化率随反应时间的变化,由于HXT 中炭的高的有序度及相对较低的环境温度(约520 ℃),导致反应前10 min 内加氢气化反应速率几乎为0。当反应进行12 min 后,HXT 中的碳开始转化。可以看出,过渡金属的催化活性明显高于碱土金属和碱金属,活性顺序为:Co ≈ Ni > Fe > > Ca ≈ Mg >K。结合图2(b)来看,过渡金属存在低温催化区域(600−750 ℃,12−18 min 的升温阶段)和高温催化区域(> 800 ℃,20 min 后的反应阶段),对应两个HXT 失重峰的出现,通过对不同温度区域转化率和失重峰的大小可以得出低温催化区域活性顺序为Co > Fe > Ni,高温催化区域活性顺序为Ni >Co > Fe,而碱金属和碱土金属只有在高温区域(>800 ℃)才能对活性炭进行缓慢的催化转化。

图2 不同催化剂对HXT 的催化加氢气化活性Figure 2 Effect of catalyst on CHG reactivity of HXT(t=850 ℃,p=3.0 MPa)(a): carbon conversion; (b): weight loss rate
关于不同碱金属、碱土金属和过渡金属催化剂的活性顺序,Yu 等[14]在800 ℃,4.0 MPa 压力下考察了10%K2CO3、10%Ca(OH)2和10%Ni(NO3)2等催化剂对呼和浩特褐煤催化加氢气化特性,发现催化剂的活性顺序为K2CO3> Ni(NO3)2> Ca(OH)2。Jiang 等[15]在800 ℃,3.0 MPa 压力下考察了2.8%K2CO3、2.8%CaO 和4.1%Ni(NO3)2等催化剂对烟煤半焦催化加氢气化特性,发现催化剂的活性顺序为CaO > K2CO3> Ni(NO3)2。文献中的不同种类催化剂活性顺序与本研究中差异的原因可能归因于反应条件的差异。已有研究表明,炭催化加氢气化过程中含碳原料的氧含量、矿物质的种类和催化剂的负载量等均对其催化加氢气化活性具有较大的影响[13,19]。Zoheidi 等[20]研究使用的原料为具有较高氧含量的低阶褐煤,碱金属催化剂在CHG 过程中易与煤中的含氧结构发生结合,形成K-O-C 结构等活性位,从而体现出高的催化加氢气化活性,而过渡金属Ni 的催化活性较低的原因可能为其负载量过高(10%),原煤的比表面积有限,大量的Ni 催化剂颗粒在反应过程极易发生团聚长大,造成催化活性降低[18]。Jiang 等[15]研究使用的原料为具有一定灰含量的烟煤半焦,其结构中的Fe 等矿物质会在反应过程中与CaO 产生协同催化作用,使得负载2.8%CaO 表现为具有较优的催化活性。因烟煤半焦的氧含量较低,负载的2.8%的K2CO3一方面,可能难以在反应过程中形成K-O-C 结构;另一方面,Zhan 等[16]研究发现,K2CO3的负载量只有达到5%以上时方可对煤展现出一定的催化加氢气化活性,因此,K2CO3表现出较低的催化活性。本研究中,在同一催化剂负载量(2%)下,以HXT为原料,可排除前人研究中的氧含量、灰分中的矿物质等因素对催化剂催化活性的干扰,得到不同种类催化剂的本征催化活性顺序。过渡金属元素在炭CHG 中展现出比碱金属和碱土金属较高的催化活性,主要归因于其在反应过程中具有较强的氢自由基供应能力和对炭结构中C−C 键的断键能力[12],由于本研究中Co 和Ni 对HXT 的整体CHG活性相近,且众多研究表明,Co 对原煤、煤焦、石油焦的催化加氢气化活性高于Ni 和Fe[18,21,22],因此,后续针对HXT 的催化加氢气化特性研究主要以Co 为催化剂展开。
2.2 Co 的负载量对HXT 催化加氢气化活性的影响
为了明确HXT 催化加氢气化过程中催化剂的最佳负载量,在氢气压力为3 MPa,温度为850 ℃的条件下,考察了Co 催化剂的负载量对HXT 催化加氢气化活性的影响。由图3(a)可知,在催化剂添加量为0.5%、1%、2%、4%和8%时,与未负载催化剂的HXT 相比,转化率得到显著提高,分别从未负载催化剂的3.39%增加至88%、94%、98%、98%和99%。从图3(b)来看,当Co 的添加量低于2%时(0.5%、1%),只有一个HXT 失重峰的出现,从HXT 开始失重对应的温度可以看到只有当反应温度高于800 ℃时,HXT 中碳才开始被催化转化。当Co 的添加量高于2%时,在反应过程中会出现两个HXT 失重峰,第一个失重峰的反应温度为600−750 ℃(低温催化区域);第二个失重峰的反应温度区间为高于800 ℃(高温催化区域),即当催化剂负载量高于2%时,反应温度达到600 ℃时,HXT 中碳即可开始被催化转化,且随着催化剂添加量的增加,更多的碳可在低温催化区域得到转化。在考察的催化剂负载量范围内,Co 的活性顺序为8%Co > 4%Co > 2%Co > 1%Co >0.5%Co。
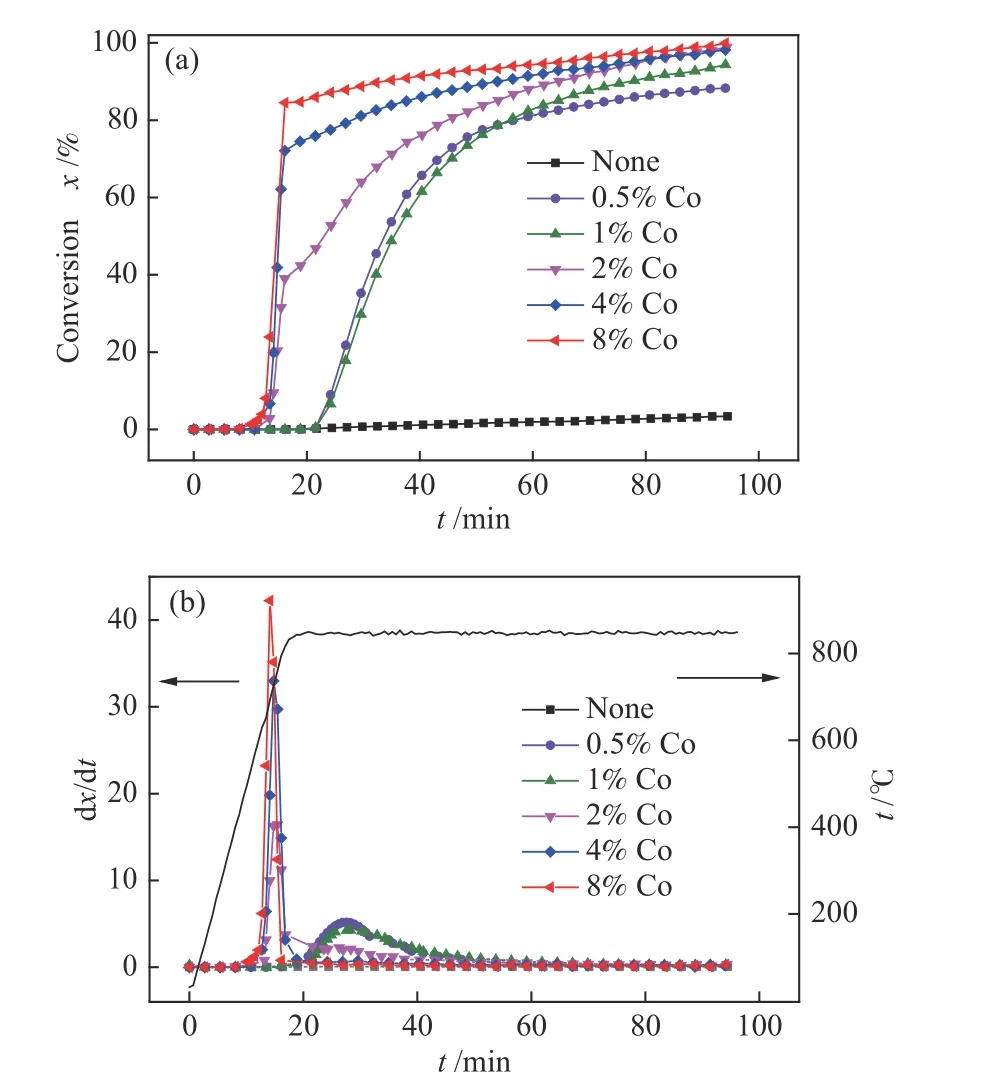
图3 不同Co 负载量对HXT 催化加氢气化活性的影响Figure 3 Effect of Co loading on CHG activity of HXT(t=850 ℃,p=3.0 MPa)(a): carbon conversion; (b): weight loss rate
关于活性炭催化加氢气化过程两个失重峰的出现,可能与两种因素有关:碳的存在形式和催化剂与碳之间的相互作用形式。Nishiyama 等[13]认为,碳物种存在一定量的反应活性较高的碳,在反应初期较低的温度下(500−700 ℃)即可被催化转化,随着反应温度的升高至800 ℃以上,半焦中缩合程度较高的碳才开始被催化转化,从而出现了两个碳失重峰。Haga 等[23]认为,两个温度区域内甲烷的生成与M-C (M:过渡金属元素)之间的不同作用形式有关。当以M的硝酸盐为催化剂时,在较低的温度区域下首先会形成M-(O)-C 结构,这种结构反应活性较高,在 400−700 ℃即可进行催化加氢气化生成甲烷,同时会伴随着氧的脱除。在较高的温度区域下(> 700 ℃),以M-C 的相互作用为主,半焦中的碳会溶解扩散到M催化剂的表面与氢自由基发生反应生成甲烷。在本研究的Co 催化HXT 加氢气化过程中,两个碳失重峰的出现的原因目前尚不清楚,为了进一步明确两个碳失重峰(特别是低温催化区域失重峰)出现的原因,需要对反应过程进行进一步分析。这将在2.4 小结进行探讨。
2.3 温度和压力对HXT 催化加氢气化活性的影响
在850 ℃下,氢气压力对负载2%Co 的HXT的催化加氢气化的影响规律如图4(a)所示。可以看到,随着反应压力从0.1 MPa 增加至1.0 MPa,HXT 的CHG 活性明显增大,当氢压进一步从1.0 MPa 增加至3.0 MPa 时,HXT 的CHG 活性未发生明显变化。在0.1−3.0 MPa 时,HXT 的转化率达到43.0%之前,碳转化规律基本一致,表明氢气压力对Co-CHG 的低温催化区域无影响。从总转化率角度来看,在0.1−1.0 MPa 升高氢气压力促进了Co 对HXT 的CHG 过程,主要是由于在较高的氢气压力可使得反应体系中H2浓度增加,H2-Co-C之间的相互作用增强,促进了Co 的供氢作用和C-H2反应,从而促进碳的转化。当压力继续从1.0 MPa 升高至3 MPa 时,整个反应过程碳的失重规律基本一致,因为碳-氢反应过程中碳的消耗对应氢的供应,表明Co 催化剂的供氢能力在1.0 MPa以上即可达到饱和,且在850 ℃,1.0 MPa 氢压以上的反应条件下,Co 催化HXT 加氢气化过程是关于氢压的零级反应。Yan 等[24]在探究压力对府谷半焦催化加氢气化过程的影响时也得到类似的结果。
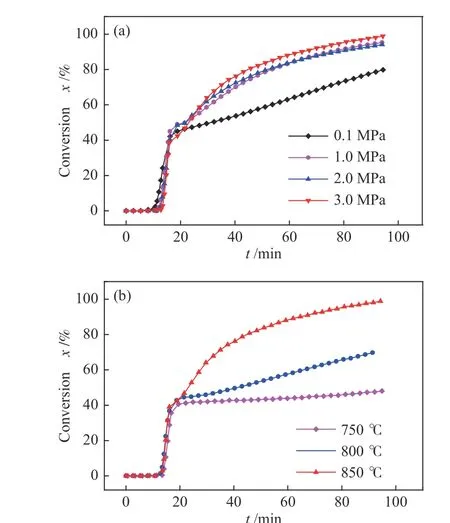
图4 压力和温度对HXT 催化加氢气化活性的影响Figure 4 Effect of pressure and temperature on CHG reactivity of HXT(a): pressure; (b): temperature
固定氢压1.0 MPa,反应温度对HXT 催化加氢气化的影响如图4(b)所示,在考察的三个温度下,低温催化区域(600−750 ℃)转化规律基本一致,当HXT 转化率达到43%后,从750−850 ℃反应温度的提升明显影响后续的碳转化过程。气化温度为750 ℃时,在达到恒定温度后,HXT 的转化率仅有小幅度增加,反应1.5 h 后从43%增加至48%。当气化温度达到800 和850 ℃后,反应1.5 h 后HXT转化率分别增加至71%和98%。可见,炭催化加氢气化过程主要为温度促使的反应。严帅的研究发现,较高的反应温度(> 750 ℃)有利于催化剂对煤中的炭结构的活化,降低反应的活化能,使之以活性较高的形式得以转化[25]。关于温度对过渡金属CHG 过程的影响,Yuan 等[26]在1 MPa 的氢气压力,650−750 ℃进行烟煤催化加氢气化,发现煤的碳转化率仍处于50%−60%。Jiang 等[8]在1.5 MPa氢气压力,在800 ℃下进行烟煤半焦催化加氢气化,发现在7.5 h 的颗粒停留时间下方可达到70%以上的转化率。以上研究中,煤/半焦催化加氢气化过程难以在短的颗粒停留时间内获取高的碳转化率,其原因主要是因为反应温度仍过于温和,若进一步提高催化加氢气化反应温度至850 ℃,则可实现在较短的颗粒停留时间下获取90%以上的碳转化率[27]。
2.4 HXT 低温催化区域出现原因的探讨
2.4.1 HXT 低温催化区域(600−750 ℃)气体产物
由图2 和图3 可以看出,在以2%Fe/Co/Ni 以及高于2%Co 负载量时,HXT 催化加氢气化过程均会出现明显的低温催化区域(600−750 ℃),为了寻求低温催化区域出现以及该区域内催化剂展现的活性差异的原因,首先利用离线色谱对不同催化剂和不同Co 催化剂负载量下在600−750 ℃生成的含碳气体产物进行定量分析,结果见图5。
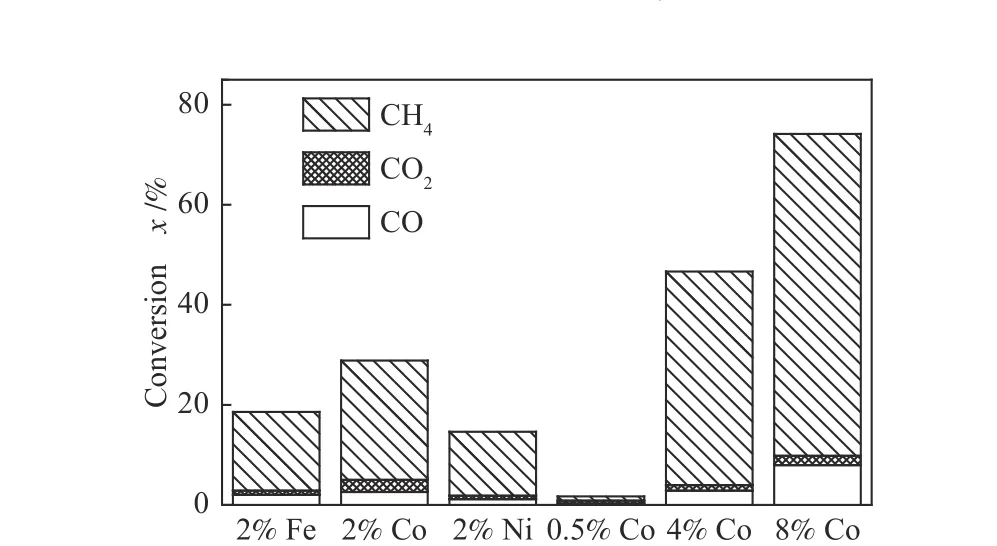
图5 HXT 低温催化区域(600−750 ℃)气体产物分析Figure 5 Analysis of gas products in low temperature catalytic zone (600−750 ℃) of HXT
从图5 可以看出,低温催化区域产物以CH4为主,同时有一定量的含氧气体(CO 和CO2)生成。通过对含氧气体中氧进行定量分析后,发现产物CO 和CO2中氧收率高于活性炭结构中1%的氧含量(表1),表明低温催化区域存在铁族金属氧化物与活性炭中碳的反应,生成了CO 和CO2。由此可见,铁族金属硝酸盐分解生成的氧化物在低温催化区域不仅能够被H2还原,而且能够与碳发生相互作用而被还原。与Fe、Ni 相比,Co 可生成更多的含氧产物和CH4,表明更多Co 的氧化物在低温反应区域被碳还原,且铁族金属氧化物与碳之间的相互作用可能在某种程度上决定了其在低温催化区域的催化活性。Co 的供氢能力要强于Fe 和Ni[12],且存在更多Co 的氧化物与碳之间的相互作用,因此,在低温催化区域过渡金属的催化活性顺序为Co > Fe > Ni。至于在高温催化区域(> 800 ℃)过渡金属的催化活性顺序为Ni > Co >Fe,可能是因为Ni 在低温催化区域转化较少的炭,因此,在较高温度转化更多的炭,这也与Tamai等[2]的研究相一致。
由图3(a)可知,当Co 负载为0.5%时,HXT 不能在低温催化区域得到转化;当Co 负载量为2%时,HXT 中40%以上的炭能够在低温催化区域得到转化;当Co 负载量进一步提高至8%时,低温催化区域得到转化的炭可达80%以上。由于0.5%Co的负载量下含氧产物生成极少,即大部分Co 的氧化物均被反应气氛中的H2还原,在低温催化区域基本不存在Co 的氧化物与碳之间的相互作用,因而较低负载量的Co 在低温催化区域未表现出催化活性。当Co 负载量提高至2%以上时,在低温催化区域同时存在Co 的氧化物不仅能够被氢气还原,而且还能够被碳还原,Co 的氧化物被碳还原的过程中可能使得HXT 中的炭结构得到活化,产生更多的活性位点,使得其低温催化区域即可被快速催化转化,从而在低温催化区域表现出较高的催化活性。Co 的负载量增大,HXT 中被活化的炭结构增多,活性位点增加,从而使得在8%Co负载量时80%以上的炭能够在低温催化区域被催化转化。
为了进一步确认负载在活性炭上的CoO 能够在低温催化区域(600−750 ℃)同时被C 和H2还原,利用HSC 热力学软件对C、CoO 和H2的反应体系进行了计算。图6 显示了在400−750 ℃时1 MPa氢压条件下,负载2%CoO(以Co 原子计负载量计)的炭进行反应后的产物分布结果。在温度低于550 ℃时,仅存在CoO 与H2的反应,生成H2O,而在温度高于550 ℃时,存在CoO 与H2和CoO 与C 的反应,生成H2O 和CO,且催着温度的升高,CO 生成量增大。图5 中的CO、CO2和CH4的生成可能主要源自于式(1)至(5)的反应。

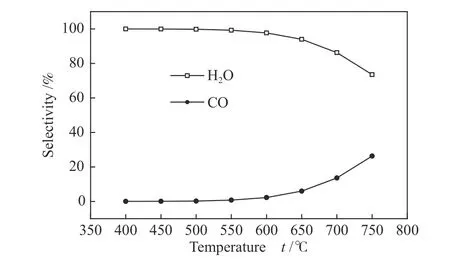
图6 负载在炭上的氧化钴被氢气和碳还原的热力学分析Figure 6 Thermodynamic analysis of the reduction of cobalt oxide loaded on carbon by hydrogen and carbon
2.4.2 负载2%Fe/Co/Ni-HXT 的TPR-MS 分析
为了实时定性检测HXT 的催化加氢气化过程产物气体析出规律,利用TPR-MS 对负载2%Fe/Co/Ni的HXT 进行原位分析,结果如图7 所示。在600 ℃左右的低温区域,Fe/Co/Ni 催化活性炭加氢气化均出现明显的CH4峰,且CH4与CO 的生成规律存在一定程度的对应关系,表明在低温反应区域铁族元素展现出的催化活性可能与其氧化物与碳的相互作用有关,铁族金属氧化物(Fe2O3、CoO、NiO 等)与碳发生反应生成CO,使得部分金属元素得到还原(提供活性氢),且部分炭结构得到活化,从而在低温区域催化活性炭加氢生成CH4。与Ni 不同的是,Co 除了在600 ℃外,在750 ℃仍存在CH4和CO 生成峰,而Ni 仅在600 ℃有一个CH4生成峰,可能是NiO 相较于CoO 而言更容易被还原[28],相应更多的Co 的氧化物与碳相互作用释放出CO(图5),同时在被碳还原过程中形成较多初始活性位点(,从而使得其在低温反应区域具有较高的催化活性。
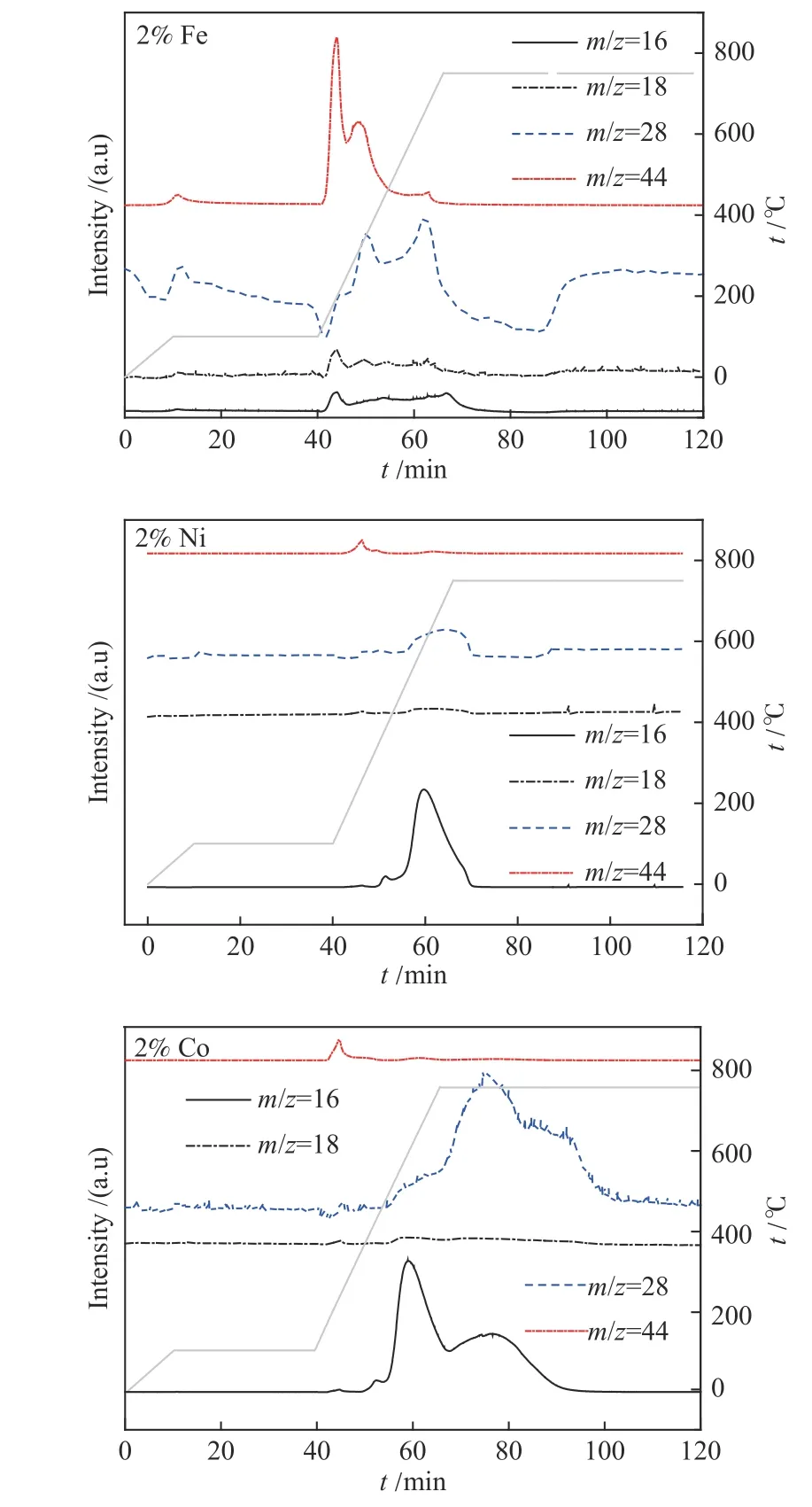
图7 负载2%Fe/Co/Ni-HXT 的TPR-MS 谱图Figure 7 TPR-MS analysis of 2% Fe/Co/Ni-HXT
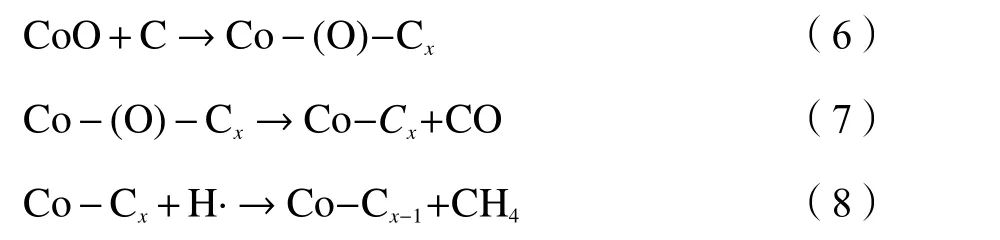
2.5 烟煤半焦/无烟煤半焦的催化加氢气化特性
前已述及,炭的CHG 过程中过渡金属催化剂在2%以上的负载量下具有较高的催化活性,且炭CHG 过程主要为温度促使的反应。但是前期工作使用的原料HXT 具有相对高的比表面积,而实际催化加氢气化过程中使用的原料为原煤/原煤半焦,其比表面积和炭结构可能与活性炭具有一定的差异,从而使得CHG 所需的反应温度和催化剂负载量可能不同。因此,需进一步考察烟煤半焦/无烟煤半焦的CHG 特性,为催化加氢实际应用过程工艺条件和催化剂负载量等因素的选择提供参考。表2 列出了四种模型炭的孔结构参数分析结果。
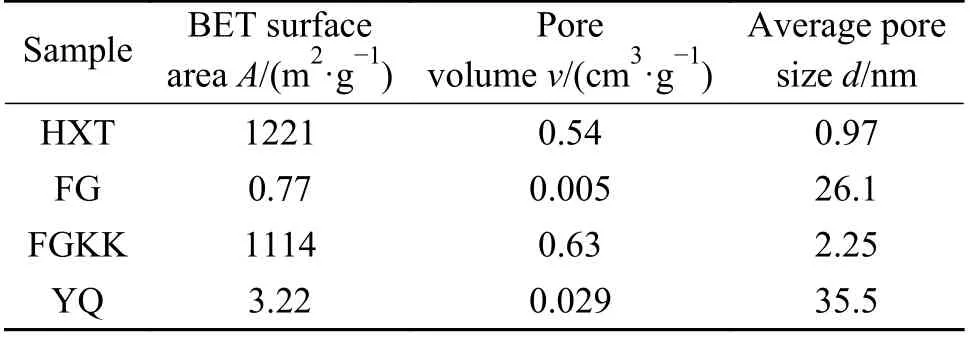
表2 不同模型炭的孔结构Table 2 Pore structure parameters of modern carbon samples
图8(a)与(b)为850 ℃,1 MPa 和不同Co 负载量下,FG 半焦和FGKK 半焦的转化率曲线,可以看出,FG 半焦在催化剂负载量为0.5%−8%时,均未出现低温催化区域,且当温度高于800 ℃时,FG 半焦中的炭方可被缓慢催化转化。反应90 min后,从0−4%Co 提高催化剂负载量时CHG 活性提高,进一步提高催化剂负载量至8%时,CHG 活性出现一定程度的降低,这主要是由于FG 半焦比表面积和孔容较低,8%Co 催化剂负载量过高可能难以均匀分散在半焦表面,导致在反应过程中催化剂易发生颗粒长大而导致活性降低的现象。在4%Co这一相对较优的负载量下,反应90 min 后转化率仅为29%。
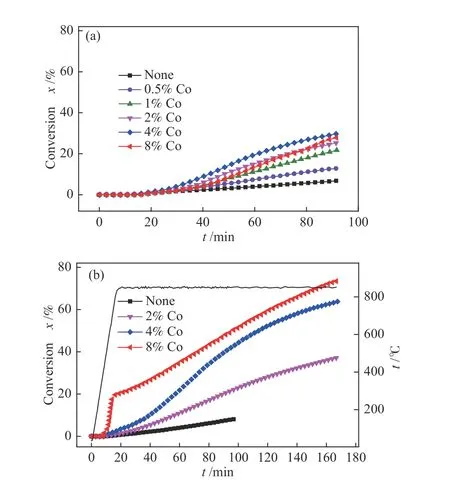
图8 模型炭的比表面积与CHG 活性的关系Figure 8 Relationship between the specific surface of model carbon and CHG activity (t =850 ℃, p =1.0 MPa)(a): carbon conversion of FG; (b): carbon conversion of FGKK
对于FGKK 半焦,从0−8%Co 提高催化剂负载量时可使CHG 活性得到提高,考察范围内最优的催化剂负载量为8%,区别于FG 半焦情况。这主要是由于FG 半焦经CO2扩孔后得到的FGKK半焦具有较FG 半焦高的比表面积,使得催化剂的最佳负载量可进一步提高。反应90 min 后,FGKK半焦在4%Co 负载量时转化率达到41%,高于4%Co负载的FG 半焦情况,说明同一催化剂负载量下,比表面积较高的FGKK 半焦具有较高的CHG 活性。当FGKK 半焦催化剂负载量进一步提高至8%时,反应3.0 h 后转化率可达73%,并出现了低温催化区域,表明比表面积较高的FGKK 半焦在一定的催化剂负载量下存在过渡金属催化剂的氧化物与碳之间的相互作用,使得部分FGKK 半焦中部分碳结构在低温催化区域得到活化并被转化。Haga 和Zhang 等[6,17]研究了负载过渡金属催化剂的不同半焦的初始比表面积对CCHG 过程的活性影响,发现过渡金属催化剂的催化活性与半焦的初始比表面积有关,较高的半焦初始比表面积有利于催化剂的分散。FGKK 半焦的比表面积大于FG 半焦,可使催化剂Co 更好的分散在炭表面,对应在相同催化剂负载量下活性位点多于FG 半焦,使得转化率得到提高。对比FG 半焦和FGKK 半焦的结果可以推测,对于比表面积较低的FG 半焦若要实现较高的CHG 活性,可在催化加氢气化过程中引入部分CO2气氛对半焦进行扩孔,进行H2+CO2气体的混合气氛CHG 可能实现低比表面积半焦的较大程度转化。
结合图8(b)与图3 来看,HXT 的比表面积和孔容与FGKK 半焦相差不大,但是HXT 负载2%Co时在90 min 内可达98%的转化率,高于FGKK 半焦负载8%Co 情况,即HXT 在较低的催化剂负载量下即可实现较高的CHG 活性。此外,FGKK 半焦在8%Co 负载量时方可出现低温催化区域,而HXT 在负载量2%Co 时即可出现低温催化区域。这表明,比表面积并非决定炭催化加氢气化活性的唯一因素,炭的CHG 活性可能还与其石墨化程度有关。HXT 具有与FGKK 半焦相近的比表面积,但其可能具有较低致密程度的炭结构,以致于其在较低的催化剂负载量下即可表现出较高的催化活性并出现低温催化区域。
以上对比研究发现,对于具有较低比表面积和较高致密程度炭结构的原料需结合炭催化加氢气化特性进一步实现其较高程度的转化。由于炭CHG 在1.0 MPa 氢压下,主要为温度促使的反应,且已有的文献研究表明,Ca 的加入能够一定程度促进过渡金属催化剂在低比表面积含碳原料表面的分散以提高其催化活性。据此,图9 给出了YQ无烟煤半焦负载4%Co 和4%Co-1%Ca 催化剂分别在850 和950 ℃的CHG 结果。可以看到,负载4%Co的YQ,从850 ℃升温至950 ℃,CHG 过程转化率得到显著提高,并且在此基础上添加1%Ca 可进一步提高4%Co 的催化活性,使得失重速率和转化率得到增加。在950 ℃,1.0 MPa 和4%Co−1%Ca 催化剂下,YQ 无烟煤半焦的转化率在反应1.5 h 后可达83.9%。因此,针对Co 催化兼具低比表面积和高缩合程度炭结构的含碳原料加氢气化过程,可通过提高CHG 温度和添加Ca 助剂的方式的实现炭CHG 反应性能的大幅提升。

图9 温度和Ca 的添加对YQ 半焦催化加氢气化活性的影响Figure 9 Effect of temperature and the addition of calcium on catalytic hydrogasification of YQ(a): carbon conversion; (b): weight loss rate (1.0 MPa)
3 结 论
对于模型炭的CHG 过程,过渡金属的催化活性明显高于碱金属和碱土金属。过渡金属催化剂存在低温催化区域(600−750 ℃)和高温催化区域(> 800 ℃);低温催化区域的出现的原因是由于过渡金属氧化物与炭发生相互作用,使得部分炭结构得到活化,从而在较低的温度下即可发生催化加氢气化生成CH4;高温催化区域主要是未被活化的炭缓慢催化加氢过程。
对于Co 催化的炭CHG 过程,在850 ℃和1 MPa氢压以上的条件下,Co 对碳-氢反应的供氢作用可达到饱和,炭的催化加氢气化主要为温度促使的催化断键反应。在同一反应温度和氢压下,炭CHG活性与比表面积、炭结构和催化剂的负载量等因素有关,比表面积越大相同催化剂负载量下炭CHG活性越高,且炭CHG 活性随催化剂负载量增大而提高。对于比表面积较低,炭结构较为致密的模型炭,可通过升高反应温度和添加Ca 助剂以大幅度提高炭CHG 活性。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
可再生能源(2022年6期)2022-06-22
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
烟台果树(2021年3期)2021-07-21
装备维修技术(2020年5期)2020-11-20
诗林(2019年6期)2019-11-14
农业与技术(2017年24期)2018-01-19
分析化学(2018年1期)2018-01-18
分析化学(2017年9期)2017-10-16
山西果树(2017年3期)2017-05-31
