
Genetic and Functional Differences of Escherichia coli Strains from Colorectal Cancer Mucosal Tissues
2022-02-16 09:20:48YuxioChngXingLiLeiDingChoYngZhiyunPnNiHnYujunCuiFhoZhiRuifuYngHongGoYujingBi
Engineering 2022年9期
Yuxio Chng, Xing Li, Lei Ding, Cho Yng, Zhiyun Pn, Ni Hn, Yujun Cui, Fho Zhi,Ruifu Yng, Hong Go*, Yujing Bi,*
a State Key Laboratory of Pathogen and Biosecurity, Beijing Institute of Microbiology and Epidemiology, Beijing 100071, China
b Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, China
c Guangdong Provincial Key Laboratory of Gastroenterology, Institute of Gastroenterology of Guangdong Province, & Department of Gastroenterology, Nanfang Hospital,Southern Medical University, Guangzhou 510515, China
d Shenzhen Center for Disease Control and Prevention, Shenzhen 518055, China
Keywords:Colorectal cancer Gut microbiota Culturomics Escherichia coli Strain level
A B S T R A C T Colorectal cancer(CRC)is the third leading cancer globally. Metagenomics has been widely used to analyze the association between the gut microbiota and CRC based on bacterial genus-or species-level comparisons,providing evidence of dysbiosis in CRC development.However,this kind of analysis is unable to provide strain-level information for understanding the individual role of a species in CRC.Here,we used culturomics to isolate CRC mucosal samples and selected 158 Escherichia coli strains to reveal their differences in both genomics and functions by means of phylogenetic analysis and inflammatory induction based on cell and animal experiments.Through genomic comparison,these strains were divided into five phylogroups.The representative strains of each phylogroup significantly induced different levels of cytokine secretion by human leukemic monocyte (THP-1 cell)-based Transwell and animal experiments.Further bioinformatic analysis revealed different profiles of single-nucleotide polymorphisms, genes,and metabolic pathways in the different phylogroups, which can improve the current understanding of the phenotypic differences between these strains. The strain differences revealed in both genomics and functions indicate that the microbiota’s function at the strain level should be investigated in order to understand the interacting mechanisms between hosts and gut bacteria.
1. Introduction
Colorectal cancer(CRC)is one of the most prevalent cancers and the third leading cause of cancer death throughout the world.Many studies have examined the association between the gut microbiota and CRC development, most of which examined sequence-based associations [1–3]. Amplicon sequencing of the 16S ribosomal RNA (rRNA) gene and metagenomic sequencing are the main methods used to study the gut microbiome,and have been widely used to identify possible pathogenic bacteria and potential diagnostic markers for CRC [4]. Once a bacterium is shown to be positively associated with CRC, animal experiments are used to verify the association. It is challenging to use DNA sequencing to determine the physiological state and function of a microorganism because the target bacteria are often purchased from a culture collection instead of being obtained as pure culture from the patients in the studies [5,6].
However, phenotypic differences occur among strains of the same bacterial species. Differences at the strain level can affect the metabolism of dietary compounds, such as galactoligosaccharides and indigestible fibers[7,8].Bacterium-mediated drug metabolism may also vary from strain to strain, affecting the efficacy and/or toxicity of drugs [9]. It is extremely difficult to accurately differentiate these features at the strain level using cultureindependent methods, although many techniques have been used to extract species- and subspecies-level information from the thousands of human-associated bacterial metagenomes available[10,11]. Culturomics is a method that uses multiple cultivation conditions for acquiring bacteria,and matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry(MS) and 16S rRNA sequencing for bacterial identification [12].This technique has become a key method in extending our understanding of the microbiome and is an important method for obtaining targeted bacterial individuals to study the mechanisms underlying the interactions between the microbiota and the host at the strain level. As shown in a recent report, Sorbara et al. [13]cultivated 273 isolates of Lachnospiraceae, including 11 genera and 27 species, from human donors and demonstrated their pathway diversities at the inter- and intra-species levels, thereby strengthening the significance of microbiota research at the strain level.
In association studies between the gut microbiota and CRC,the samples are predominantly derived from feces, which are easy to obtain for further analyses. Intestinal mucosa-associated bacteria might play more important roles in the interaction between the microbiota and the host. Several studies have investigated the mucosal microbiota associated with CRC using 16S ribosomal DNA (rDNA) amplicon sequencing [14,15], and have found that the diversity and composition of the gut microbiota in the mucosa differ from those of the microbiota in feces. A study noted that intestinal mucosal samples may be better suited for use in determining the relationship between the microbiota and the host[16]. In order to further demonstrate the differences at the strain level of mucosa-associated bacteria, we selected 158 strains of Escherichia coli(E.coli)isolated from 44 intestinal mucosal samples of 22 patients with CRC(from both the tumor mucosa and the adjacent normal mucosa) in our culturomics research project of mucosa-associated bacteria, for whole genomic sequencing using next-generation sequencing technology and for cytokine induction at both the cell and animal level. Next, the virulence-associated gene distribution and metabolic pathways were further analyzed to identify the possible mechanisms of functional differences. We confirmed that the different strains of E.coli did indeed show genomic variations and functional differences.
2. Materials and methods
2.1. Samples
The 22 CRC patients included in the study were from Beijing Shijitan Hospital. The inclusion criteria for these volunteers were:①being CRC patients aged 50–80 years; ②having no previous chemoradiotherapy; and ③meeting the criteria for surgical treatment.The CRC mucosal tissue was 2 cm2of CRC tissue sampled at Beijing Shijitan Hospital, fully ground in 2 mL of phosphatebuffered saline (PBS). The tissue-related bacteria were isolated and cultured under the predetermined culture conditions.
2.2. Culturomics
The bacteria were isolated and identified using culturomics. In brief, the mucosal tissue was placed in sterile PBS for grinding.All samples were cultured under basic culture conditions (aerobic or anaerobic, 37 °C, yeast casitone fatty acids (YCFA) solid culture medium,no preincubation).When colonies appeared on the plates,individual colonies were picked and transferred to liquid culture enrichment medium in 24-well plates. The enrichment cultures were frozen in glycerol and used to inoculate solid medium for detection. Each colony was then analyzed with an Autof ms1000 spectrometer (Autobio, China). If the colonies could not be accurately identified with MALDI-TOF MS, the isolate was identified with 16S rRNA gene sequencing. The 16S rRNA gene was sequenced by Tsingke Biological Technology Company (China).
2.3. Bioinformatics analysis
2.3.1. Classification of E. coli
The E.coli genome was sequenced by Novogene Biological Technology Company (China). The sequence data have been uploaded to the GenBank (BioProject PRJNA608078). We used the Clonal-Frame software [17] for the analysis and classification of E. coli,based on the sequences of eight housekeeping genes(4095 nucleotides in total) [18]. We used the Snippy pipeline v4.3.8†† https://github.com/tseemann/snippy.‡ http://treesoft.sourceforge.net/treebest.shtml.for singlenucleotide polymorphism (SNP)calling,with E. coli strain K-12 substrain MG1655 (accession number: U00096) as the reference genome. SNPs located in repetitive regions were excluded from the phylogenetic analysis. Repetitive regions were identified with tandem repeats finder (TRF) v4 and self-alignment by nucleotide basic local alignment search tool (BLASTn). The genome-wide SNPs were used to construct a neighbor-joining tree using the TreeBest software‡† https://github.com/tseemann/snippy.‡ http://treesoft.sourceforge.net/treebest.shtml., and all the phylogenetic trees were visualized with ggtree.
2.3.2. Analysis of E. coli genes
We performed the de novo assembly with SPAdes. The assemblies were subjected to gene annotation with Prokka, and the annotation results (GFF3 files) were used in Roary to identify the pan-genome and to generate the gene presence/absence matrix.We used eggNOG-Mapper to annotate the clusters of orthologous groups (COG) classification of the pan-genes.
2.4. Transwell experiment
A Transwell plate is a nest that divides the pores into the upper and lower chambers. The membrane in the middle of the nest can have various pore sizes. We chose a pore size of 0.4 μm to ensure that the bacterial products could pass through the membrane but the bacteria could not. Human leukemic monocytes (THP-1 cells)were plated in the lower chambers of 12-well Transwell plates(Thermo Fisher Scientific,USA)at a seeding density of 1×106cells per well (1 × 106cells∙mL-1) in Roswell Park Memorial Institute(RPMI) 1640 medium (Thermo Fisher Scientific). Each Transwell apparatus was then incubated with 100 ng∙mL-1phorbol 12-myristate 13-acetate (PMA) (Thermo Fisher Scientific) for 48 h at 37 °C under humidified air containing 5% carbon dioxide (CO2) to stimulate the differentiation of the cells into macrophages. Each strain of E. coli was centrifuged to remove the bacterial medium and then suspended in cell culture medium. Finally, the different strains of E.coli were added to the upper chambers of the Transwell plates (1 × 107per well) at a multiplicity of infection (MOI) = 10.RPMI 1640 medium alone was used as the negative control. After incubation for 3 h at 37 °C under humidified air containing 5%CO2, the cell culture medium was collected. The THP-1 cells were purchased from BeNa Culture Collection Company (USA), and the cell culture medium was RPMI 1640 supplemented with 10%bovine serum (Cleson Scientific, China).
2.5. Animal experiment
Female BALB/c mice (eight weeks of age) were purchased from Vital River Laboratory Animal Technology (China). The mice were randomly divided into five groups of five mice each: A1, A2, D1,D2,and blank control(BL).After the mice had adapted to the environment for one week, broad-spectrum antibiotics were added to their drinking water: 1 g∙L-1ampicillin, 1 g∙L-1neomycin sulfate,1 g∙L-1metronidazole,and 0.5 g∙L-1vancomycin[19].Immediately after feeding,each mouse was gavaged with 1×109E.coli twice a week for four weeks, and blood samples were taken from the tail once a week.The blank control group was given the same amount of sterile PBS.
2.6. Cytokine measurements
The culture supernatants of the THP-1 cells incubated in the Transwell apparatus with E. coli or medium alone were harvested and assayed with the Bio-Plex 200®system (Bio-Rad, USA).
2.7. Analysis and statistics
A pairwise comparison of the cytokine contents in multiple groups was performed.The normality and homogeneity of variance of each set of data were first confirmed.If the data were distributed normally and showed homogeneity of variance, analysis of variance (ANOVA) was used, whereas if they were not normally distributed or showed heterogeneity of variance, a rank sum test was used(p ≤0.05).Pearson’s χ2test was used to compare the distributions of pathogenic genes between the two groups. The total sample numbers of the two groups were defined as n and their theoretical frequency as T. When n ≥40 and all T ≥5, Pearson χ2test was used,but when n<40 or T<1,Fisher’s exact probability test was used (p ≤0.05).
2.8. Ethics
Ethics approval and consent to participate were obtained from each volunteer,and the research was approved by the Ethics Committee of Beijing Shijitan Hospital (2018KY55).
3. Results
3.1. E. coli strains from CRC mucosa
Mucosa is the most relevant tissue for intestinal diseases. We isolated and cultured 44 mucosal samples using culturomics methods.E. coli was the most prevalent species isolated from both cancer and adjacent mucosa. It was isolated from all 44 samples(Fig.S1 in Appendix A). All E. coli strains and patients’information are shown in Table S1 in Appendix A.
3.2. Genetic diversity of E. coli in the gut mucosal microbiota of CRC patients
The genetic structure of commensal E. coli is determined by a variety of host and environmental factors. The factors that determine its virulence may reflect its adaptation to the symbiotic environment. We selected 158 strains of E. coli from both tissues for whole-genome sequencing. Among these strains, those from the same patient were employed to access the within-host diversity,and others from different patients were used for comparisons between hosts. The software ClonalFrame was used to classify and analyze the E. coli [17,20]. The strains were divided into five phylogroups:A,D,B1,B2,and F(Fig.1).However,the E.coli strains from the cancer tissues and adjacent tissues were not clearly distinguished on the phylogenetic tree, indicating that there were no genotypic differences between the populations from the two types of tissues.
3.2.1. Within-host diversity
A previous study has shown that,at any one time,a person usually carries a dominant microbiota strain that accounts for more than half of the colonies isolated, while other strains are present at different levels [21]. Here, we noted that some patients carried a dominant phylogroup. For example, in patient 18, most strains belonged to phylogroup B1;however,they could be further differentiated into different sub-branches based on SNP analysis, indicating differences in these strains at the genomic level. In patient 2, only strains from phylogroup A were isolated; in contrast, in patient 3, the strains were assigned to three phylogroups, without a dominant phylogroup.
3.2.2. Between-host diversity
Bacterial diversity at the strain level between hosts is high;as in line with our estimation,we did not detect a single strain shared by any two patients. These results further emphasize that an understanding of a bacterial strain’s function is critical in in precisely identifying bacterial mechanisms in disease development. Therefore,we employed immune cell and animal experiments to further evaluate the cytokine induction effect of the representative strains from different phylogroups.
3.3. Effects of different E. coli strains on THP-1 cells
Because the gut microbiota does not come into contact with immune cells directly in the normal state, the effects of the gut bacteria on immune cells are mainly mediated by their metabolites. Therefore, a Transwell method was used to investigate the effects of different E. coli strains on immune cells [22]. A total of 36 representative strains (from six patients) were selected for the experiments according to the phylogroup classification. A Transwell plate was divided into upper and lower chambers,and THP-1 cells were added to the lower chamber. We induced the cells with PMA for 48 h to promote their differentiation into macrophages, and then added the corresponding E. coli strain to the upper chamber (MOI = 10). After incubation for 3 h, the cell culture medium was collected to measure the levels of interleukin (IL)-1β, tumor necrosis factor α (TNF-α), and IL-6(Fig. 2(a)).
First, we compared the effects of E. coli strains from different phylogroups on the secretion of these three cytokines by the macrophages (Figs. 2(b)–(d)). The results showed differences among the five phylogroups. The amounts of cytokines secreted after treatment with phylogroup D strains were lower than those with other phylogroups,and those with phylogroup A strains were always higher than those with phylogroup D. We also examined whether the sample source and sample classification affected macrophage cytokine secretion. The results suggested that there were significant differences between the different phylogroups, even when the strains were from the same patient;however,the cytokine-inducing effect was similar within the same phylogroup, even if the samples were from different patients(Figs.2(e)–(g)).In summary,the phylogroup of a strain had a more obvious effect on cytokine secretion by macrophages than the source of the strain.
3.4.Effects of different E.coli strains on gut–microbiota-depleted mice
Because there were significant differences between the effects of phylogroups A and D in the cell experiment, we selected two strains of E. coli from each phylogroup for analysis in an animal experiment. We designated the four strains as A1, A2, D1, and D2,respectively,and the BL group.We first fed the mice with combined antibiotics for one week. After the antibiotic treatment, the mice were gavaged twice a week for four weeks with the corresponding E.coli strain,and blood samples were collected two days after the second gavage each week (Fig. 3(a)). No differences in body weight were observed among the groups during the experiment (Fig. S2 in Appendix A). At the end of the experiment,compared with the control group, there was no significant change in colorectal pathology in the four experimental groups (Fig. S3 in Appendix A). The contents of IL-1β, TNF-α, and IL-6 in the sera were detected once each week (Figs. 3(b)–(d)). In week 1, the changes in the cytokines were consistent with the results of the cell experiment, but no differences were observed among the four experimental groups and the control group in weeks 2 and 3.However, in week 4, the concentrations of cytokines in the sera were inconsistent with those in week 1, insofar as they were significantly higher after treatment with phylogroup D than after treatment with phylogroup A.
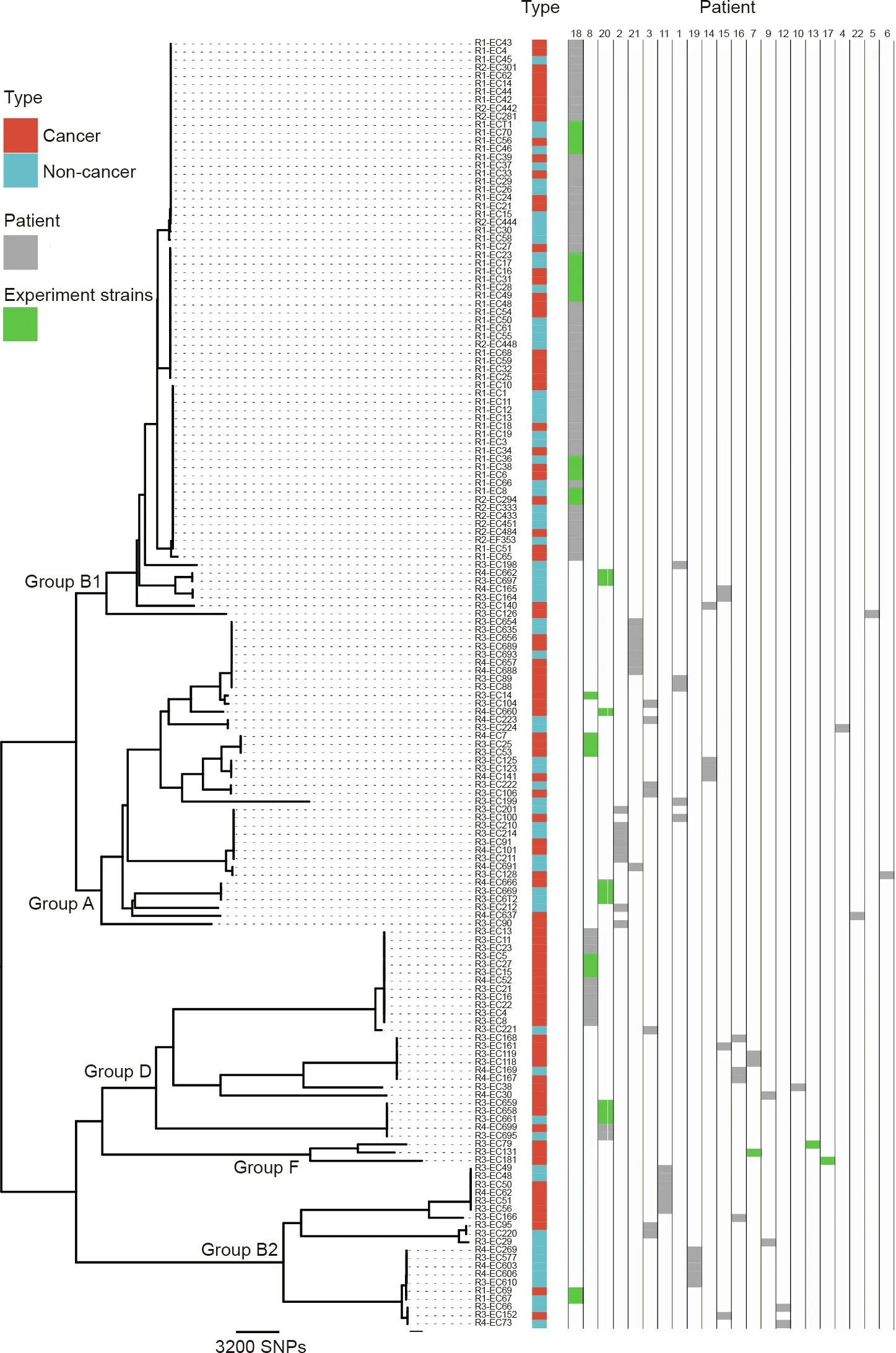
Fig.1. Phylogenetic tree of CRC-related E.coli.158 strains of E.coli from 22 patients were analyzed.These strains can be grouped into five main phylogenetic groups:A,B1,B2,D,and F.Red indicates strains from cancer tissues and blue indicates strains from non-cancer tissues.Further Transwell experiment strains are shown in green shading.
3.5. Genetic analysis of E. coli strains in phylogroups A and D
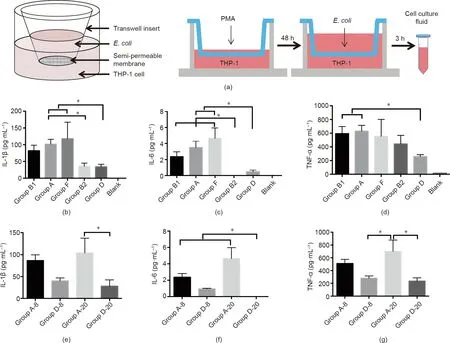
Fig. 2. (a) Design of Transwell experiments. (b) Comparison of IL-1β content in cell culture medium among groups. (c) Comparison of IL-6 content in cell culture medium among groups. (d) Comparison of TNF-α content in cell culture medium among groups. (e) Within-group comparison of IL-1β content in cell culture medium. (f) Withingroup comparison of IL-6 content in cell culture medium. (g) Within-group comparison of TNF-α content in cell culture medium. *p ≤0.05.
In order to explore the possible explanations for the cytokineinducing differences between the two E. coli phylogroups in mice,we further analyzed the genomic differences of the 14 strains used in the cell experiment(six from phylogroup D and eight from phylogroup A) for virulence-associated gene distribution and metabolic pathways. We found 33 248 SNPs in the lineage-specific core genes and 348 in the lineage-specific accessory genes between phylogroups A and D(Fig.4(a)and Table S2 in Appendix A).When we mapped the SNP-related genes to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, 37 pathways differed between the two phylogroups (Table S3 in Appendix A). Among the 348 genes, 278 were specifically present in phylogroup D and 70 in phylogroup A. Most of these genes were related to bacterial metabolism, and a small number were virulence-associated genes related to cell invasion and adhesion. The pathways associated with the core genes included amino acid,nucleotide,carbohydrate,and inorganic ion transport and metabolism.The pathways related to the accessory genes involved only carbohydrate transport and metabolism (Fig. S4 in Appendix A)
In E. coli, more than 100 genes are reportedly related to virulence [23–31]. We screened 103 virulence-associated genes or genomic islands and compared these genes in the phylogroup A and D strains. We then calculated the proportions of these genes present in each phylogroup (Table S4 in Appendix A). The significantly different genes are shown in Figs. 4(b)–(d), which shows all the known differential genes in the sequenced strains (Fig.4(b)), the strains used in the cell experiment (Fig. 4(c)), and the strains used in the animal experiment (Fig. 4(d)). Fourteen genes(chuA, ipaH, fmlA, fimA, fimC, ecpA, ecpD, kpsS, ydeR, FimH, iss, aufC,
lpfA, and stfD) were more frequent in phylogroup D than in phylogroup A (p ≤0.025), and 12 genes (yfcV, ChiA, elfC, stcD, papC,
After everyone had gone, my mother told me, You want to be the same as American girls on the outside. She handed me an early gift. It was a miniskirt in beige tweed. But inside you must always be Chinese. You must be proud you are different. Your only shame is to have shame.
iucC, iucD, iutA, iha, sat, papF, and papGII) were more prevalent in the strains of phylogroup A than in those of phylogroup D(p ≤0.050). Several genes in phylogroup D (e.g., chuA, ipaH, and fmlA) are reportedly linked to adhesion and invasion, and yfcV in phylogroup A is related to adhesion.
Although we did not find any of the same strains in different patients,we observed the same virulence gene profiles in the strains from different patients(Fig.S5 in Appendix A).For example,strains R4-EC223 and R3-EC224, from patients 21 and 4, respectively,shared the same known virulence gene profile.In another example,strains R3-EC201 and R3-EC100,which were not only from different patients,but also from different mucosal sites(cancer and adjacent tissues),also shared the same virulence gene profile.
4. Discussion
Most of the literature focusing on intestinal bacteria and CRC has employed metagenomics to identify the associated bacteria,followed by the use of strains from a culture collection center,such as the American Type Culture Collection (ATCC), for further verification. For example, high-virulence Fusobacterium nucleatum was found to be associated with CRC patients, and strains ATCC 23726 and 25586 were used to induce tumors in germ-free mice[5,32]. Although a tumor was successfully induced after intestinal mucosa damage by azoxymethane(AOM),it is better to employ the original strain from CRC patients for the validation experiments,which may be of great help in understanding the pathogenic role of the target bacterium in cancer development.
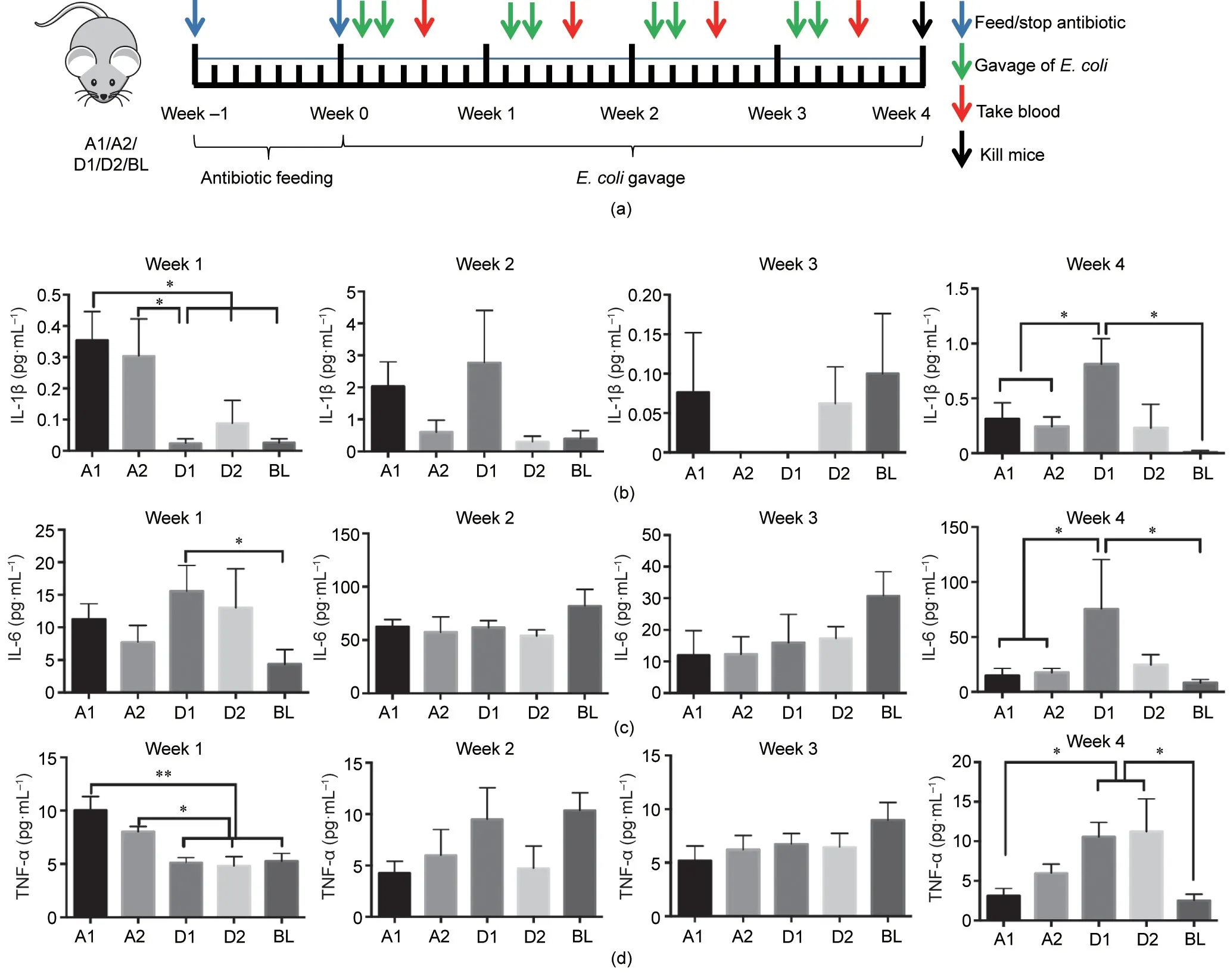
Fig.3. (a)Design of mice experiments.(b)Comparison of IL-1β content in mouse sera from week 1 to week 4 after gavage.(c)Comparison of IL-6 content in mouse sera from week 1 to week 4 after gavage. (d) Comparison of TNF-α content in mouse sera from week 1 to week 4 after gavage. *p ≤0.05; **p ≤0.01.
In this study, E. coli strains isolated from the mucosa tissue of CRC patients were used as a model to show strain differences in both genome and function. E. coli are common bacteria in the human intestine, some of which cause gastrointestinal diseases[33] while others cause urinary system [34] and nervous system infections [35]. Metagenomic data have also suggested the presence of multiple E. coli strains in individual patients with Crohn’s disease [36]. According to the genomic classification scheme, 158 strains of E. coli were assigned into five phylogroups: A, D, B1,B2,and F.We found that the E.coli strains from individual patients belonged to different phylogroups, indicating high diversities of bacterial strain in the gut microbiota of different hosts. Next, we were interested in finding out the functional differences of these phylogroups.
Cytokines are important indices reflecting the immune responses of tumor patients. IL-1β, TNF-α, and IL-6 are three cytokines whose expression changes significantly in cancer, and their levels can even be related to the stage of tumor development[37,38]. Therefore, using Transwell experiments, we first quantified the secretion of these three cytokines by macrophages in response to different E. coli strains from the various phylogroups.The differences among different phylogroups were obvious, and significant differences were detected between the levels of these cytokines induced by phylogroups A and D. We noted that strains from a single patient belonged to different phylogroups. Do these strains differ in their induction of cytokine secretion by immune cells? The strains in phylogroups A and D from patients 8 and 20 were compared. There was no difference in the induction of cytokines by E. coli strains from the same phylogroup. However, the levels of cytokines secreted by immune cells clearly differed when induced by the different phylogroups of E.coli(e.g.,phylogroups A-20 and D-20 from the same patient induced different levels of IL-1β and TNF-α secretion). These results suggest that the intestinal microbiota should be studied at the strain level in order to obtain an accurate understanding of the role of a bacterial strain in the gut.
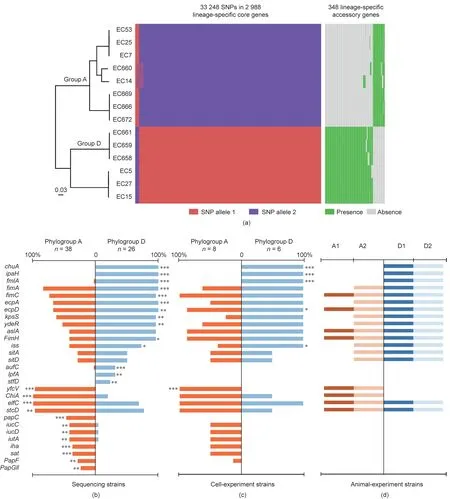
Fig. 4. (a) Comparison of SNPs in the lineage-specific core genes and lineage-specific accessory genes between phylogroup A and phylogroup D. (b)–(d) Distribution of pathogenic genes in phylogroups A and D:(b)all sequencing strains;(c)only strains used for the cell experiment;and(d)only strains used for the mice experiment.Numbers indicate the ratio of the number of strains carrying genes to the total number of strains from each phylogroup. Genes presenting statistically significant differences are depicted. *p ≤0.05; **p ≤0.01; ***p ≤0.001.
We then challenged mice with strains from phylogroups A and D to see if similar results could be detected in an animal model.Before the bacterial challenge, all the mice were treated with antibiotics to clear their gut microbiota [19]. After gavage with the target bacteria for one week, the serum levels of IL-1β and TNF-α in the mice differed significantly between phylogroups A and D, which was consistent with the results of the cell experiment. There were no differences in the serum cytokine profiles of the two mouse groups challenged with different phylogroups over the next two weeks.However,in week 4 after gavage,the strains of phylogroup D again caused significantly greater cytokine secretion in the mice than those of phylogroup A. This is an interesting phenotypic result, which we sought to explain at the gene level.We first analyzed the present and absent genes in phylogroups A and D.In total,352 phylogroup-specific accessory genes were identified, which were mostly present in phylogroup D. In phylogroup A, most of the E. coli strains were enterotoxigenic E. coli (ETEC),which cause disease by attaching to the host’s epithelial lining via surface proteins, called colonization factors (CFs), and possibly other surface structures [23]. However, adherent-invasive E. coli(AIEC), which are associated with inflammatory bowel disease and can produce toxins that cause intestinal inflammation and lesions [27], were predominantly found in phylogroups D and B2.Studies have shown that E. coli expressing the yfcV gene, which is an adhesion gene that encodes the major subunit of a putative chaperone-usher fimbria, are more likely to adhere to the mucosa than those that do not, causing inflammation [39]. Expression of the yfcV gene was significantly higher in phylogroup A than in phylogroup D,which may be related to the significantly elevated cytokine expression in phylogroup A in the cell experiment and in the mouse sera in the first week after the challenge.However,with the extension of infection time, phylogroup D, which has more virulence genes than phylogroup A, may induce greater cytokine production in the host. It was also noted in another study that AIECs use a variety of virulence factors to promote their invasion of cells [40].
Researchers have reported that different bacterial strains affect human health differently, but the available isolates and genome collections of most bacterial species in the human gut are still limited,especially in terms of strain-level diversity.Furthermore,most of the strains used for strain-level research come from a large number of individuals, and our knowledge of the diversity of strains from a single human host is very limited. Some studies have pointed out functional differences between strains of the same species, which could affect human health [44,45]. Here, we used culturomics to isolate mucosa-associated bacteria from the tumor mucosa and its adjacent tissues in CRC patients, and provided a CRC-related gut microbial bank for future functional studies.Sequencing of E. coli and function evaluation of E. coli strains on the cell and host clarified the possible mechanism (Fig. 5), which provided in-depth screening, analysis, and verification of the bacteria at the strain level.
Acknowledgments
This research was supported by the National Natural Science Foundation for Key Programs of China (81790632), the National Natural Science Foundation of China (31970863), and the Innovation Leader Team Program of Guangzhou (201809010014).
We thank Janine Miller,Ph.D., from Liwen Bianji,Edanz Editing China (http://www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Authors’ contribution
Ruifu Yang conceived the study;Ruifu Yang,Yujing Bi,and Hong Gao designed the experiments;Yuxiao Chang did the experiments and analyzed the data;Xiang Li did the experiments;Lei Ding collected the samples and analyzed the data;Chao Yang did bioinformatics analysis; Zhiyuan Pan and Ni Han did the experiments and investigated the literature; Yujun Cui and Fachao Zhi directed the experiments; and Yuxiao Chang, Yujing Bi, and Ruifu Yang wrote and revised the manuscript.
Compliance with ethics guidelines
Yuxiao Chang, Xiang Li, Lei Ding, Chao Yang, Zhiyuan Pan, Ni Han, Yujun Cui, Fachao Zhi, Ruifu Yang, Hong Gao, and Yujing Bi declare that they have no conflict of interest or financial conflicts to disclose.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.eng.2021.03.028.

- Engineering的其它文章
- Innovative Scanners Boost Medical Imaging Quality and Utility
- Building a Highly Stable Ultrathin Nanoporous Layer Assisted by Glucose for Desalination
- Introduction of a New Method for Regulating Laves Phases in Inconel 718 Superalloy during a Laser-Repairing Process
- Mesoscopic Modeling Approach and Application for Steel Fiber Reinforced Concrete under Dynamic Loading: A Review
- Effects of Medicines and Supplements on Spontaneous Pregnancy and Semen Parameters in Male Infertility: A Systematic Review Update and Network Meta-Analysis
- Applications of CyTOF in Brain Immune Component Studies