
ZnO对于CaO吸附CO2性能影响的DFT研究
2022-02-16 13:52张友浩徐运飞王菲菲李英杰
中南大学学报(自然科学版) 2022年12期
张友浩 ,徐运飞 ,王菲菲 ,李英杰
(1.山东大学 能源与动力工程学院,山东 济南,250061;2.山东大学 高效节能及储能技术与装备山东省工程实验室,山东 济南,250061)
为了尽快实现“双碳”目标,解决环境问题与能源问题,研究CO2高效捕集与分离技术具有重要意义。目前,CO2捕集方法主要有固体吸附剂、碱性液体洗涤、离子液体吸收等化学方法以及金属有机框架材料(MOFs)吸附、沸石吸附、膜分离等物理方法[1-3]。其中,CaO基吸附剂的碳酸化/煅烧循环反应捕集CO2是一种具有前景的大规模脱碳技术[4]。因该方法具有能耗低、成本低等优点,目前已经应用于燃烧后捕集CO2[5-6]、热化学储能[7-8]、吸收CO2强化制氢[9-11]等技术中,有望在未来广泛应用于工业生产。
然而,天然CaO 基吸附剂如石灰石、白云石等吸附CO2性能随着循环次数增加而降低。采用掺杂添加剂对天然CaO 基吸附剂进行改性是提高其吸附CO2性能的有效方法之一[8]。LI等[12]发现MnO和SiC能够提高CaO吸附CO2的性能,且两者同时添加时对于CaO 的CO2吸附性能具有协同促进作用,当吸附剂内CaO,MnO 和SiC 的质量比为 100∶5∶5 时,改性CaO 吸附CO2性能最佳;SUN等[13]向CaO 中掺杂CeO2,发现掺加了1%(质量分数)CeO2的CaO 碳酸化转化率达到0.94,比未改性CaO 提高了35%。WANG等[14]以电石渣为CaO 前驱体制备了多种Ni/CaO 吸附剂,发现Ni/CaO 的CO2吸附率显著高于天然CaO 吸附剂的CO2吸附率。此外,使用Na,Co,Fe,Zr和La等金属氧化物对CaO 进行改性,提高其CO2吸附性能,这也被广泛研究[15-17]。
ZnO 是良好的导热材料,ZnO 掺杂能够提升CaO 的导热性能,有利于CaO 吸附剂的再生过程[18-19]。目前ZnO 已作为添加剂用于提升天然钙基材料的CO2吸附性能[19-20]。LIU等[20]制备了 ZnO改性CaO吸附剂(CaO/ZnO),研究了ZnO添加量和预处理温度对于CaO/ZnO 吸附CO2性能的影响,发现当CaO 与ZnO 的物质的量比为8∶1 时,CaO/ZnO的吸附CO2性能最佳,相比于未改性CaO提升23.8%。
然而,目前ZnO 掺杂对于CaO 吸附CO2性能的研究停留在宏观实验层面,对于微观原子层面的机理尚不清楚。因此,有必要通过模拟计算从微观原子层面揭示ZnO 对CaO 吸附CO2性能的影响机理。本文通过挤出成型法制备ZnO 掺杂CaO吸附剂(ZnO/CaO),研究ZnO/CaO 的CO2吸附性能。基于密度泛函理论(DFT)模拟计算,建立了CaO(0 0 1)和Zn-CaO(0 0 1)表面模型,计算2 种材料吸附CO2的构型、吸附能、分态密度以及差分电子密度,从微观原子层面揭示ZnO 掺杂对CaO 吸附CO2性能的影响机理。
1 实验系统与计算方法
1.1 实验样品制备
通过等体积浸渍法制备ZnO/CaO。为便于实际工业应用,通过挤出成型法将吸附剂挤压成型[21]。相比于CaO,Ca(OH)2具有更好的黏性,更易于成型,是一种常用的制备成型钙基材料的原料[21-23]。而醋酸盐可在高温煅烧后形成金属氧化物,且释放出的气体会使材料具有更好的孔隙结构,是应用广泛的添加剂前驱体[23]。因此,采用Ca(OH)2和醋酸锌(ZnC4H6O4)(AR,天津市科密欧化学试剂有限公司)作为原材料,制备成型的ZnO掺杂CaO吸附剂。
ZnO 掺杂CaO 吸附剂(ZnO/CaO)制备流程如下:
1) 向一定质量的ZnC4H6O4中加入蒸馏水并充分搅拌至完全溶解,获得醋酸锌溶液,其中ZnC4H6O4的质量通过吸附剂内ZnO 质量分数计算得到;
2) 向溶液中加入150 g Ca(OH)2,充分浸渍后置于干燥箱内,在80 ℃下干燥12 h 得到ZnO/ Ca(OH)2粉末;
3) 向ZnO/Ca(OH)2粉末内加入质量分数为3%的聚乙烯吡咯烷酮作为黏结剂,加入少量蒸馏水,充分搅拌至吸附剂形成柔软膏体,然后利用液压机挤出成型;
4) 将获得的ZnO/Ca(OH)2材料在空气中干燥48 h 后,移至干燥箱内,在80 ℃的条件下干燥8 h;
5) 充分干燥后,将吸附剂置于马弗炉内,在850 ℃下煅烧15 min,得到ZnO/CaO吸附剂。
KHOSA等[19]研究发现,添加质量分数为5%的ZnO/CaO 具有最佳的储热性能。因此,为探究添加ZnO 对于CaO 吸附CO2的作用,制备ZnO 的质量分数分别为1%,3%和5%的ZnO/CaO。为了比较ZnO 掺杂的效果,采用上述步骤(不加入ZnC4H6O4)制备了成型CaO吸附剂。
1.2 实验流程
利用加压双固定床系统进行ZnO/CaO 吸附CO2性能测试实验。加压双固定床系统如图1 所示。由图1可见:利用加压固定床作为碳酸化反应器,常压固定床作为煅烧反应器。向常压固定床通入3 L/min 的N2,设定反应器温度为880 ℃。加压固定床内通入CO2,并迅速将压力提升至 0.2 MPa,设定反应器温度为850 ℃。
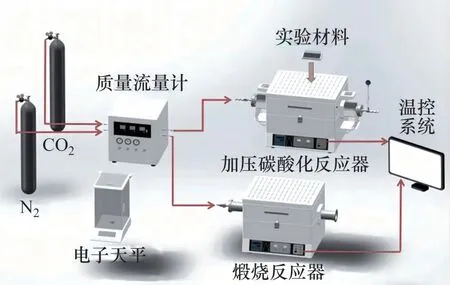
图1 加压双固定床系统Fig.1 Pressurized double fixed bed system
实验流程如下:取5 g ZnO/CaO样品置于瓷舟内,将样品送入碳酸化反应器,密封反应器后增大CO2流量进行加压,在0.2 MPa 和850 ℃的条件下反应15 min后取出样品,在干燥皿(充入N2)内冷却4 min 后称得反应后样品质量。称质量完成后,将样品送入煅烧反应器内,在纯N2、880 ℃的条件下煅烧20 min 后取出样品,置于干燥皿内冷却 4 min 后称质量。以上即为1 次碳酸化/煅烧吸附CO2循环。称质量完成后,将样品再次送入碳酸化反应器,进行下一次循环。实验条件的设定均基于前期研究[21]。
利用碳酸化转化率评价ZnO/CaO 的吸附CO2性能。XCaCO3,N表示吸附CO2的CaO 质量与样品内CaO总质量之比,计算式如式(1)所示。
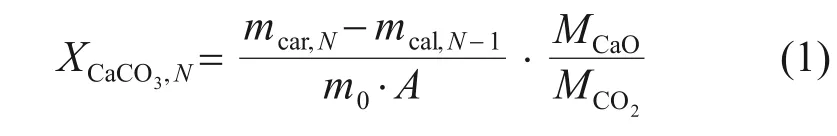
式中:XCaCO3,N为第N次循环后ZnO/CaO 的碳酸化转化率;mcar,N为第N次碳酸化后的样品质量,g;mcal,N-1为第N-1次煅烧再生后的样品质量,g;m0为样品初始质量,g;A为ZnO/CaO 内ZnO 的质量分数,%;MCaO与MCO2分别为CaO 与CO2的摩尔质量,g/mol。
1.3 计算方法
基于密度泛函理论(DFT)的量子力学方法被普遍认为是一种揭示反应微观机理的有效途径。目前,DFT 计算已经被普遍应用于研究添加剂增强CaO吸附CO2机理。近年来,CaO基材料CO2吸附机理的DFT研究总结如表1所示[12,24-28]。
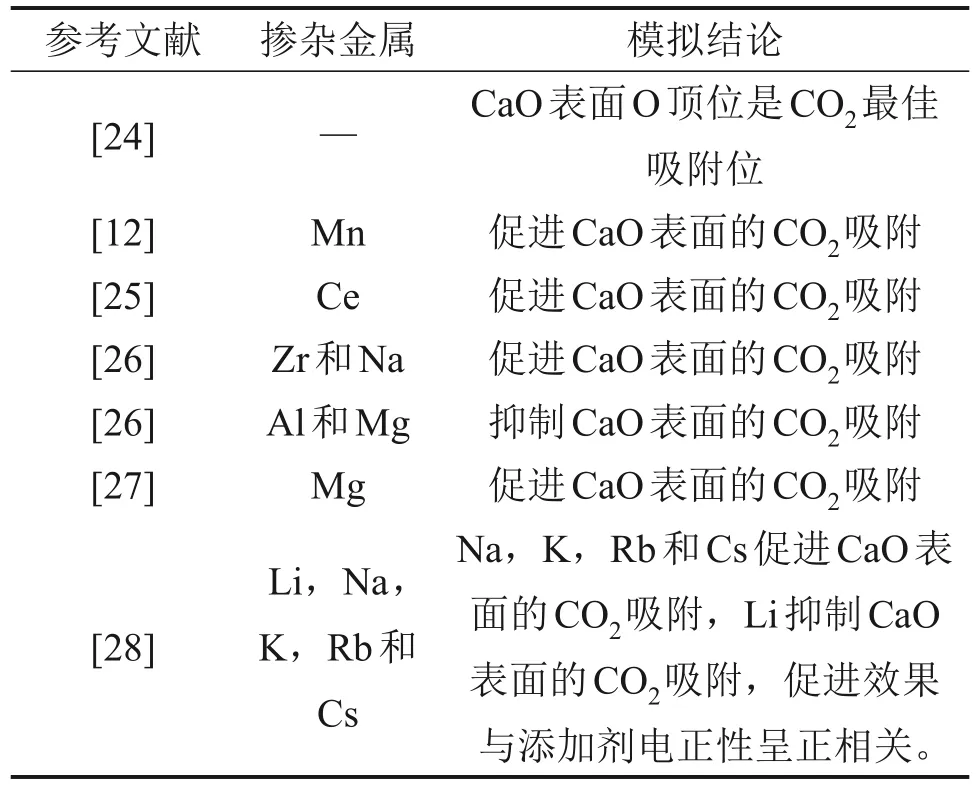
表1 基于DFT计算的掺杂改性对CaO吸附CO2的影响机理[12,24-28]Table 1 CO2 adsorption mechanism of CaO-based adsorbent based on DFT calculation reported in literatures[12,24-28]
为了从原子层面揭示Zn 掺杂对于CaO 吸附CO2性能的影响机理,通过CASTEP程序,采用非限制自旋的Generalized-Gradient-Approximation (GGA)和Perdew-Burke-Ernzerhof (PBE)作为交换相关泛函进行了DFT 计算。基元的原子能量、作用力、压强和位移的收敛容差分别设置为5×10-6eV,0.1 eV/nm,0.02 GPa 和5×10-5nm。采用BFGS优化算法,电子截断能设置为630 eV,采用4×4×2的布里渊区的k点取样,采用OTFG ultrasoft赝势。
原子替换法是利用添加剂金属原子替换基底内金属原子以形成掺杂添加剂基底模型的方法,是通过DFT 计算材料添加金属氧化物时常用的模型构建方法,YAN等[25]利用原子替换法构建了 Ce-CaO(0 0 1)表面,探究了Ce 掺杂和预先吸附H2O对于CaO基吸附剂CO2吸附性能的影响,计算结果与实验结果相符合;WANG等[29]利用Ni 替换Ca 构建了Ni-CaO(0 0 1)表面,研究了Ni/CaO 的强化制氢性能;LI等[12]利用原子替换,构建了 Mn-CaO(0 0 1)表面,探究了Mn 对CaO 基吸附剂CO2吸附性能的影响。
研究表明,CaO(0 0 1)表面的表面能为0.647 J/m2,是CaO能量最低、最稳定的表面[30-31],因此,选取CaO(0 0 1)表面作为模型进行计算。以CaO(0 0 1)表面为基础,通过原子替换法构建了Zn-CaO(0 0 1)表面模型进行计算。利用吸附能Ead评价ZnO/CaO材料表面CO2吸附能力,如式(2)所示。
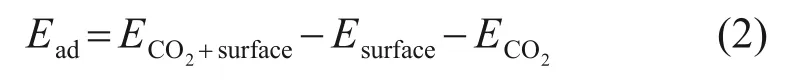
式中:ECO2+surface为CaO 基吸附剂吸附CO2后的系统总能量,eV;Esurface为材料表面能,eV;ECO2为CO2分子的能量,eV。Ead为负值,说明材料表面与CO2之间相互吸引作用较强,Ead越小,说明吸附过程越容易发生;Ead为正值,说明材料表面与CO2分子间相互排斥,吸附过程很难发生或不吸附,Ead越大,说明越难以发生吸附。
2 结果与讨论
2.1 ZnO添加量对于CaO吸附CO2性能的影响
不同ZnO 添加量的ZnO/CaO 在5 次循环中的碳酸化转化率如图2 所示。从图2可见:随着循环反应次数增加,ZnO/CaO的碳酸化转化率下降,但相同循环次数时ZnO/CaO 的转化率比CaO 的转化率更高;随着ZnO 添加量从1% 增加到5%,ZnO/CaO的循环碳酸化转化率不断增大,但当ZnO添加量超过3%时,对于ZnO/CaO的碳酸化转化率变化不大。因此,3%的ZnO添加量是合适的。
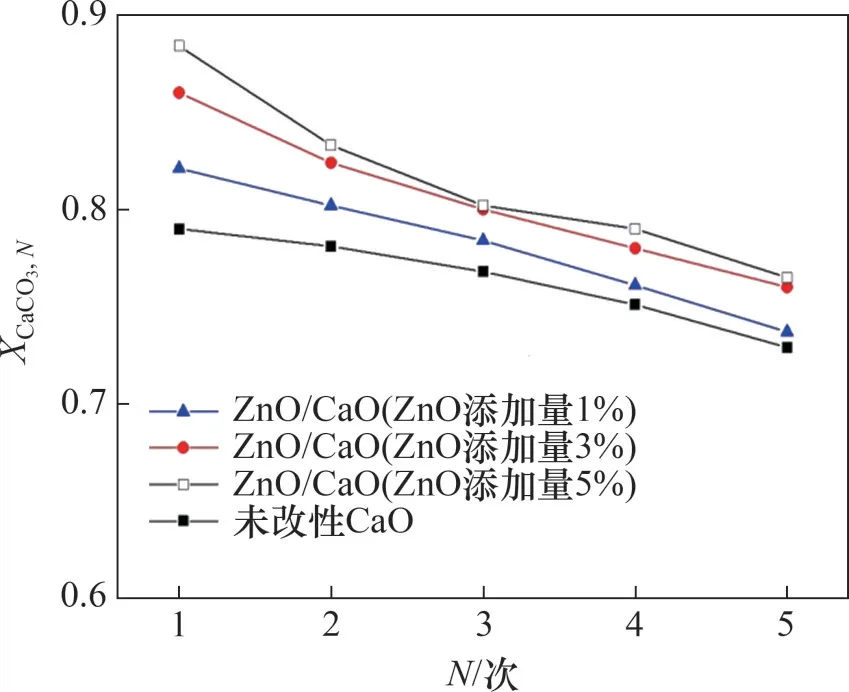
图2 ZnO添加量对于CaO在多次循环中吸附CO2性能的影响Fig.2 Effect of ZnO addition on CO2 adsorption performance of CaO in multiple carbonation/calcination regeneration cycles
2.2 Zn-CaO(0 0 1)表面吸附CO2构型与吸附能
建立CaO(0 0 1)表面三维周期性结构表面,保留3层原子,其中,底层原子固定,表面2层原子及CO2分子弛豫。其中,建立2×2的超晶胞以更好地展示CaO 表面特性,并在模型上方建立高度为1 nm 的真空层以防止结构周期性对计算结果的影响。
将CO2模型置于边长为2 nm的立方晶体中进行几何结构优化。采用原子替换的方式构建Zn- CaO(0 0 1)表面,将CaO(0 0 1)表面顶层的1个Ca原子替换为Zn 原子,优化结构后获得Zn-CaO(0 0 1)表面模型。O顶位已经被证实是CaO表面CO2最佳吸附位[24]。因此,选择O 顶位作为CO2吸附位点(对应O原子称为Oβ),CO2初始位置设置在Oβ原子正上方垂直距离0.3 nm处。优化结构后获得CO2在CaO(0 0 1)表面及Zn-CaO(0 0 1)表面最佳吸附构型。
CaO(0 0 1)表面与Zn-CaO(0 0 1)表面吸附CO2前后的最佳构型分别如图3 和图4 所示。由图3 可以发现:相比于CaO(0 0 1)表面,Zn-CaO(0 0 1)表面Zn原子处明显出现塌陷,且O原子向Zn原子方向发生细微偏移。计算结果表明,Zn—O 键长度为0.237 nm,低于Ca—O 键长度(0.241 nm)。这说明Zn原子与O原子之间的化学键比Ca原子与O原子之间更强,键能更高,更强的化学键也导致 Zn-CaO(0 0 1)表面相比于CaO(0 0 1)表面结构更加稳定。吸附CO2后CaO(0 0 1)表面的Ca 与Zn-CaO (0 0 1)表面的Zn都向远离Oβ处偏移,但Zn偏移更为明显。这是由于Oβ在吸附CO2前,仅与Ca和Zn之间存在电子转移;而吸附CO2后,Oβ与C发生相互作用成键,使得Oβ从C 处获得部分电子,导致Oβ与Ca 或Zn 间的电子转移数减少,而Zn 与O 间电子转移量变化更大,导致Oβ与Zn间相互作用力变化更大,因而使得Zn发生明显偏移。
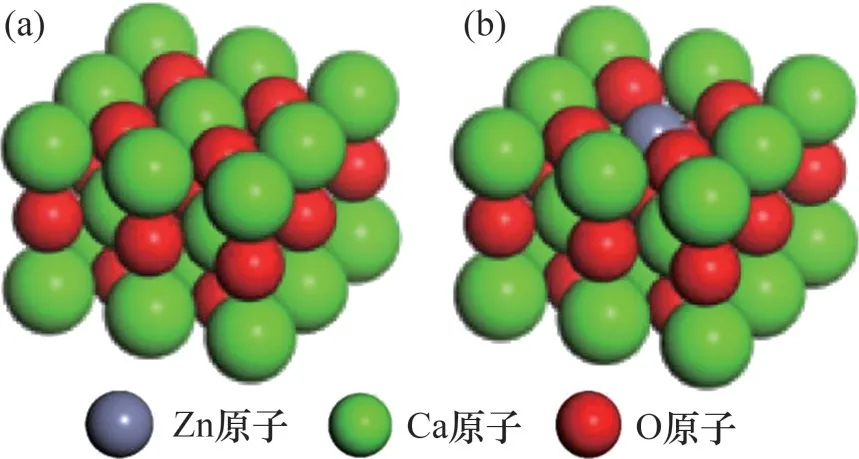
图3 CaO基材料表面模型示意图Fig.3 Surface models of surfaces of CaO-based materials
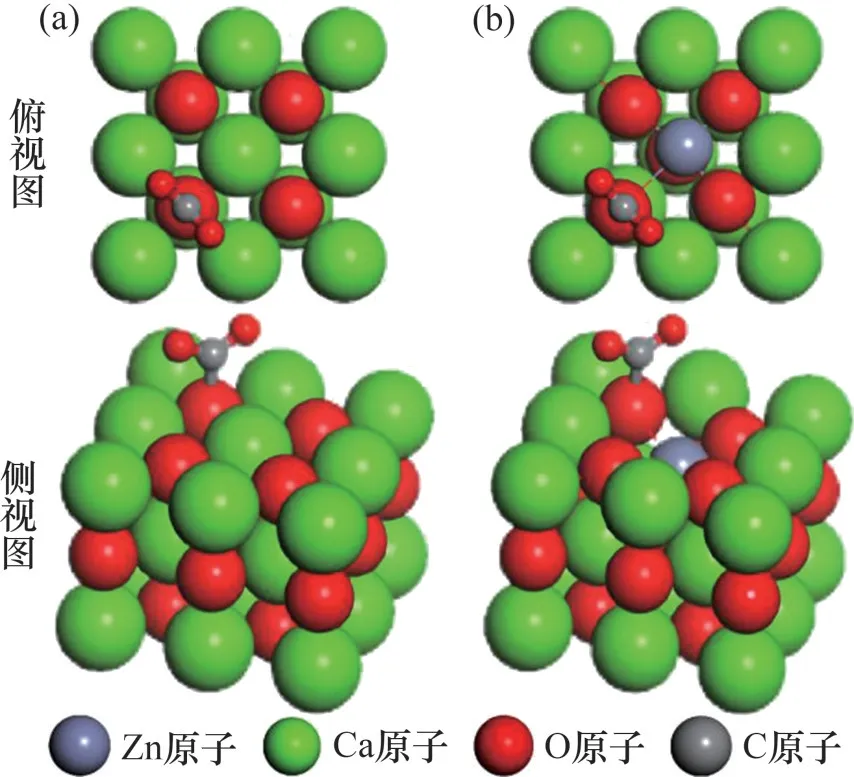
图4 CaO基材料表面吸附CO2模型示意图Fig.4 Models of CO2 adsorbed on surfaces of CaO-based materials
表面吸附CO2构型及吸附能的模拟计算结果如C—O 键长分别为0.142 nm 与0.141 nm,CO2内 C—O 键长均为0.126 nm,键角分别为128.96°和128.63°。因此,CaO(0 0 1)与Zn-CaO(0 0 1)表面吸附CO2后的结构较为一致,且均与的结构类似[32],这说明CaO(0 0 1)与Zn-CaO(0 0 1)表面对于CO2均是通过化学反应进行吸附,CO2与表面O原子结合,以结构吸附在材料表面。在吸附能方面,CaO(0 0 1)表面的CO2吸附能为-1.184 eV,而加入Zn 后,Zn-CaO(0 0 1)表面CO2吸附能降低至-1.947 eV,这表明掺杂ZnO 显著提升了CaO 的CO2吸附性能,Zn-CaO(0 0 1)对于CO2的吸附能力更强,这也与实验所得结论相符合。
2.3 Zn-CaO(0 0 1)表面吸附CO2的电子特性
电子态的变化可以由CO2吸附前后的分态密度(Projected Dos,PDOS)图像来揭示。当PDOS图内2 个原子电子轨道出现共振峰时,说明2 个原子间有化学键生成,具有强烈的电子相互作用[33]。图5所示为CaO基材料表面吸附CO2前的PDOS图。由表2 所示。由表2 可见:CaO(0 0 1)表面与Zn-CaO(0 0 1)表面吸附CO2后,表面Oβ原子与C原子之间图5 可见:在吸附CO2前,Zn-CaO(0 0 1)表面Zn原子与O 原子之间出现了更多的共振峰。这表明了相比于Ca原子,Zn原子与O原子之间电子转移量更多,成键能力更强,结构更为紧密,因此,加入Zn 提高了CaO 表面的结构稳定性。此外,与CaO(0 0 1)表面相比,Zn-CaO(0 0 1)中O 原子的态密度峰更低且跨度更大,这表明Zn-CaO(0 0 1)表面中O 原子的离域性更强,Zn 原子掺杂导致O 原子被活化,更易与吸附质发生反应。
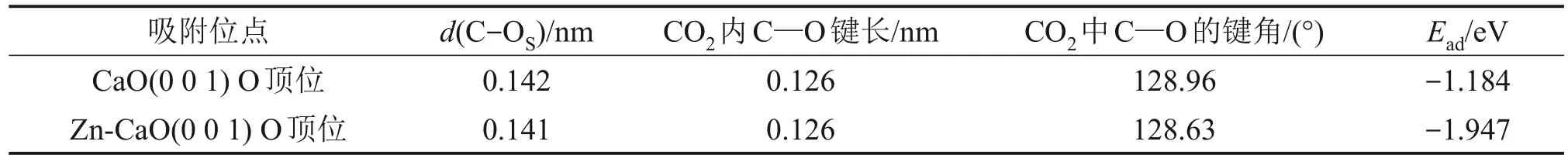
表2 CaO(0 0 1)与Zn-CaO(0 0 1)表面吸附CO2的几何参数与吸附能Table 2 Geometrical parameters and adsorption energy of CO2 on CaO(0 0 1) and Zn-CaO(0 0 1) surfaces
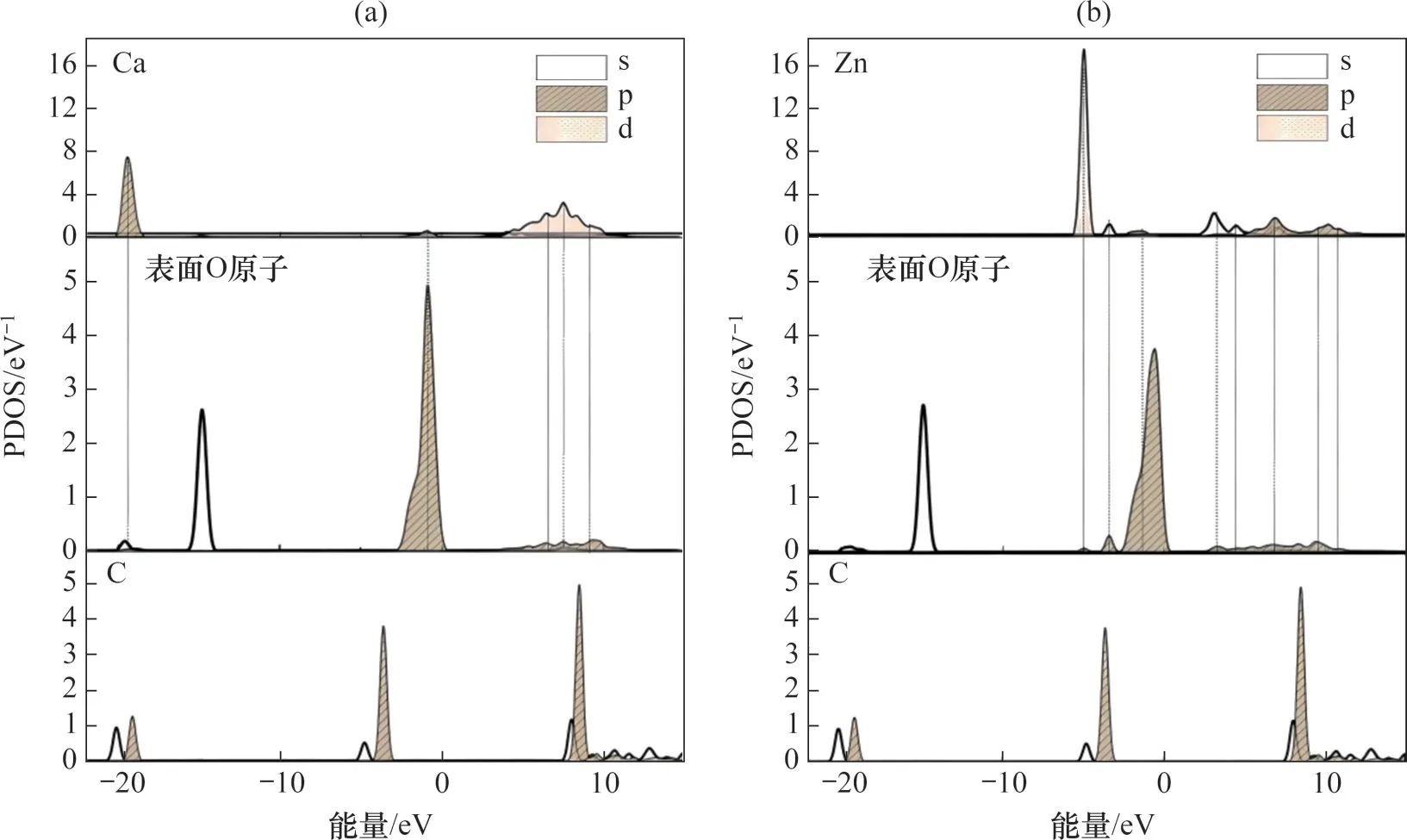
图5 CaO基材料表面吸附CO2前的PDOS图Fig.5 PDOS of surfaces of CaO-based materials before adsorption of CO2
吸附CO2后CaO(0 0 1)表面与Zn-CaO(0 0 1)表面模型内各原子PDOS图如图6所示。由图6可见:相比于吸附CO2前,吸附CO2后CaO(0 0 1)表面与Zn-CaO(0 0 1)表面Oβ原子与C 原子之间均明显呈现出杂化现象,出现大量共振峰。这表明在CaO(0 0 1)表面与Zn-CaO(0 0 1)表面内C原子与Oβ原子均发生强烈的相互作用。相比于吸附前,吸附后的C原子与O原子的态密度峰均明显向低能区发生偏移,这表明CO2吸附形成的CO32-结构更加稳定,CO2得以稳定吸附在钙基材料表面。此外,吸附CO2后,Ca原子和Zn原子与C原子和Oβ原子之间均出现3个原子电子轨道共振峰,这说明在CO2吸附过程中,部分电子由Ca原子、Zn原子转移至Oβ原子将O原子激活,进而转移至C原子。
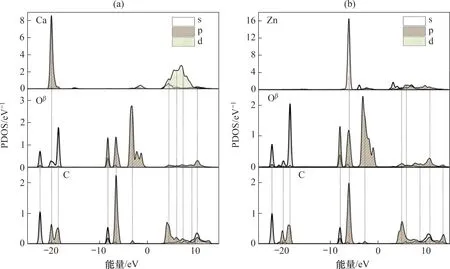
图6 CaO基材料表面吸附CO2后的PDOS图Fig.6 PDOS of surfaces of CaO-based materials after adsorption of CO2
相比之下,Zn-CaO(0 0 1)表面的Zn 原子与C原子、Oβ原子在-6.06 eV处出现高态密度的3个原子电子轨道共振峰,而CaO(0 0 1)表面内Ca 原子与C原子、Oβ原子的3个原子共振峰普遍较小,态密度较小。这说明Ca 原子与C 原子、Oβ原子之间的电子转移量较少,相互作用较弱,而Zn 原子与C 原子和Oβ原子之间电子转移数量明显偏高。Zn原子与Oβ原子之间更多的电荷转移导致表面Oβ原子更容易被激活,进而通过与C原子的电子相互转移吸附CO2,Zn与Oβ原子之间的相互作用是ZnO/CaO具有更强CO2吸附性能的关键。
CaO(0 0 1)表面与Zn-CaO(0 0 1)表面内Ca,Zn和O 原子之间的电子转移情况分别通过差分电子密度(electron density difference,EDD)图像表示,如图7 所示,吸附在表面的CO2及表面Oβ原子的EDD图像如图8所示。由图7可以发现:相比于Ca原子,Zn 原子与表面O 原子之间的电子转移量更多,这表明Zn原子和表面上的O原子之间存在强相互作用。图8 中,CaO(0 0 1)表面及Zn-CaO(0 0 1)表面Oβ原子与C 原子的电子云出现了明显重叠,这表明Oβ原子与C 原子之间发生了强烈的电子相互作用并形成化学键。相比于CaO(0 0 1)表面,Zn-CaO(0 0 1)表面吸附的CO2内C原子与表面O原子的电子转移量更多,电子相互作用更加强烈。因此,Zn原子的电子损失导致Zn原子周围的电子不饱和,这有利于电子从邻近表面的CO2转移,并促进CO2与Zn-CaO 表面的键合。差分电荷密度和分态密度分析结果都表明,添加ZnO 后吸附剂具有更高的结构稳定性,且ZnO 掺杂引起的电子转移提高了表面的反应活性。
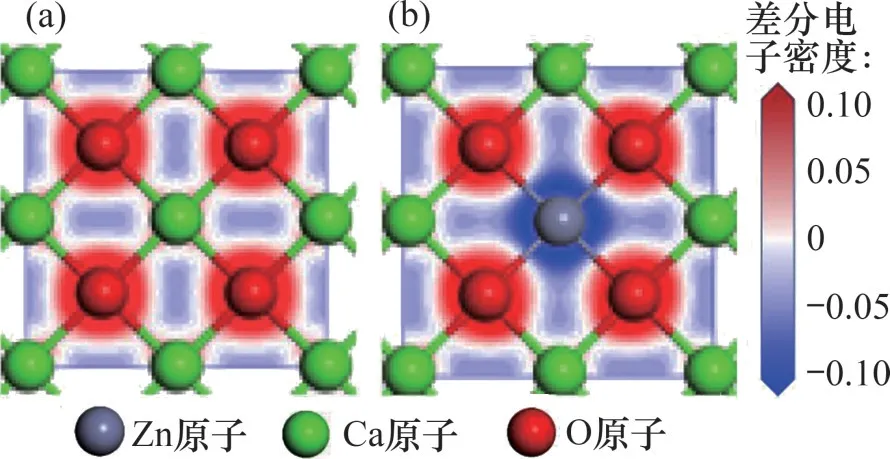
图7 CaO基材料表面顶层EDD图Fig.7 EDD of top layer of surfaces of CaO-based materials
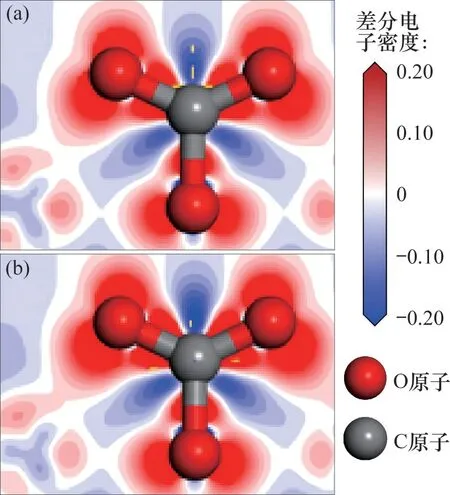
图8 CaO基材料表面吸附CO2的EDD图Fig.8 EDD of CO2 adsorbed on surfaces of CaO-based materials
3 结论
1) 添加ZnO增强了CaO的吸附CO2性能,在5次碳酸化/煅烧循环中,相同循环次数时ZnO/CaO的碳酸化转化率始终高于纯CaO。当ZnO 添加量为1%~5%时,随着ZnO添加量增加,ZnO/CaO的循环碳酸化转化率不断增大,但当ZnO 添加量超过3%时,ZnO/CaO的碳酸化转化率变化不大,因此,3%是较合适的添加量。
2) CaO(0 0 1)与Zn-CaO(0 0 1)表面CO2吸附构型与吸附能计算结果显示,Zn 原子与O 原子之间的相互作用力显著强于Ca 原子与O 原子之间的相互作用力,导致Zn-CaO(0 0 1)表面结构更加稳定。同时,加入Zn导致CaO(0 0 1)表面的CO2吸附能由-1.184 eV降低至-1.947 eV,促进了CaO吸附CO2。
3) Zn与O之间的电子转移量明显高于Ca原子与O 原子的电子转移量,且ZnO/CaO 内O 原子的反应活性更高。Zn 原子、Oβ原子和C 原子在 -6.06 eV处存在高态密度的3个原子电子轨道共振峰。Zn 原子与O 原子之间的电子转移导致O 原子离域性增强,进而更容易与CO2反应成键,有利于CO2在CaO 吸附剂表面的吸附。Zn,Oβ和C 之中的电子在3 种原子内连续转移,Zn 和Oβ原子之间的电子转移是ZnO/CaO 具有更强CO2吸附性能的关键。
猜你喜欢
化工管理(2022年13期)2022-12-02
机械工业标准化与质量(2022年6期)2022-08-12
能源工程(2022年1期)2022-03-29
小天使·二年级语数英综合(2021年5期)2021-07-11
能源工程(2021年1期)2021-04-13
矿产综合利用(2020年1期)2020-07-24
天然气化工—C1化学与化工(2018年6期)2018-02-20
中学化学(2017年2期)2017-04-01
中国调味品(2017年2期)2017-03-20
中国塑料(2016年9期)2016-06-13
