
超高效液相色谱-四级杆/静电场轨道阱高分辨质谱联用快速测定水产品及干制水产品制品中的116种农药和24种生物毒素残留
2022-02-15 07:58王勇张宪臣华洪波李云松黄碧嘉李勇
现代食品科技 2022年1期
王勇,张宪臣*,华洪波,李云松,黄碧嘉,李勇
(1.中山海关技术中心,广东中山 528403)(2.卓雅外语学校,广东中山 528401)
随着人民生活水平的提高,水产品及其制品在人们生活水平中的比重日益增大,水产品及其制品安全问题成为全社会关注的焦点。在影响水产品及其制品质量安全的众多因素中,农药和生物毒素残留是主要的原因之一,果蔬和粮食等作物在生产和加工过程中使用农药以后,其残留可能经食物链和环境传递而进入到水产品及其制品中,进而影响人体健康[1,2]。农药主要是指用来防治危害农林牧业生产的有害生物和调节植物生长的化学药品[3,4]。生物毒素是一类由生物自身分泌代谢、半生物合成的对其他生物物种有毒害作用的各种化学物质,通常以痕量形式存在于食品基质中,对人类的膳食健康造成了巨大的潜在威胁[5,6]。
水产品及其制品中农药和生物毒素残留受到广泛的关注,许多国家和国际组织均制定了相关的残留限制标准。日本从2006年5月29日实施的《食品中残留农药的肯定列表制度》中对涉及水产品的134种化学药物残留量限定[7]。美国FDA对水产品进口要检查221种化学药物的残留,严禁使用的药品有10种[8]。欧盟、加拿大、韩国等国家或地区近几年也相应提高了水产品及其制品中农药和生物毒素残留的限制[9]。
水产品及其制品中农药和生物毒素检测方法主要有酶联免疫法[10-12]、气相色谱-串联质谱法[13-15]、液相色谱-串联质谱法[16-21]和液相色谱-高分辨质谱法[22-26]。其中酶联免疫法易出现假阳性结果;气相色谱-串联质谱法需对样品进行繁琐的衍生化处理;液相色谱-串联质谱法有较好的选择性、灵敏度和特异性,但是其定性准确度方面尚有欠缺。水产品及其制品中农药和生物毒素检测的前处理方法包括固相萃取、快速溶剂萃取、微波辅助萃取、分子印迹技术和QuEChERS等[22-28]。QuEChERS由于具有快速、简单、便宜、有效、耐用和安全可靠等优势在检测水产品及其制品中农药和生物毒素应用日趋增多,但是其易产生基质效应,会对方法的检测限、选择性,以及测试结果的准确定量产生影响。
四极杆/静电场轨道阱高分辨质谱仪(Q-Exactive)具有分辨率高及定量能力好的优点,可以利用目标物母离子的精确分子量直接定量,无需对目标物逐个优化子离子及相关参数,对于多目标物分析可以极大地降低检测方法的时间,同时又能很好地避免低分辨质谱易受基质干扰而产生假阳性的现象[29-32]。
本研究利用 QuEChERS方法对水产品及其制品前处理,选择一种新型的高效基质脂肪吸附剂(EMR-Lipid)对样品杂质进行吸附,能够有效的降低基质效应。使用超高效液相色谱-四级杆/静电场轨道阱质谱法(UPLC-Q Exactive Orbitrap MS)对水产品中116种农药和24种生物毒素残留量进行测定,方法灵敏、快速、简单、准确、省时、稳定、实用性强,适用于食品实验室大批量检测水产品样品。
1 材料与方法
1.1 材料、试剂与仪器
黑鱼、罗非鱼和罗氏虾由中山海关石歧办查验一科提供,干制鲈鱼、干制鲍鱼等样品购自中山华润万家超市。
标准品:甲萘威(纯度≥98.9%)、呋喃丹(纯度≥98.3%)、3-羟基克百威(纯度≥98.0%)、异丙威(纯度≥98.5%)、亚胺硫磷(纯度≥98.5%)、敌百虫(纯度≥99.0%)、啶虫脒(纯度≥98.1%)、氯磺隆(纯度≥98.5%)、炔草酯(纯度≥98.0%)、炔草酸(纯度≥99.0%)、霜脲氰(纯度≥99.0%)、野燕枯(纯度≥99.0%)、烯唑醇(纯度≥99.0%)、克瘟散(纯度≥99.0%)、仲丁威(纯度≥97.7%)、唑螨酯(纯度≥99.4%)、抑霉唑(纯度≥99.0%)、精甲霜灵(纯度≥98.7%)、多效唑(纯度≥99.0%)、伏杀磷(纯度≥99.0%)、辛硫磷(纯度≥98.0%)、抗蚜威(纯度≥98.7%)、敌稗(纯度≥99.0%)、哒螨灵(纯度≥99.0%)、戊唑醇(纯度≥98.5%)、噻菌灵(纯度≥98.89%)、噻虫啉(纯度≥98.5%)、脱叶膦(纯度≥95.0%)、三环唑(纯度≥97.0%)、咪鲜胺(纯度≥99.0%)、多菌灵(纯度≥99.0%)、噻螨酮(纯度≥98.0%)、地虫磷(纯度≥94.5%)、嘧霉胺(纯度≥99.0%)、苯酰菌胺(纯度≥98.5%)、嘧菌环胺(纯度100.0%)、氯苯嘧啶醇(纯度≥99.0%)、苯线磷亚砜(纯度≥98.5%)、苯胺磷砜(纯度≥99.0%)、氟虫腈砜(纯度≥98.2%)、甲草胺(纯度≥99.5%)、毒死蜱(纯度≥99.0%)、乐果(纯度≥98.5%)、倍硫磷(纯度≥98.0%)、氟硅唑(纯度≥99.5%)、氟胺氰菊酯(纯度≥94.0%)、茚虫威(纯度≥98.5%)、稻瘟灵(纯度≥99.0%)、对硫磷(乙基对硫磷)(纯度≥99.5%)、甲基嘧啶磷(纯度≥99.0%)、喹硫磷(纯度≥96.0%)、吡螨胺(纯度≥99.5%)、四氟醚唑(纯度≥99.6%)、唑啶磷(纯度≥97.8%)、丁酮砜威(纯度≥91.3%)、噻虫胺(纯度≥98.9%)、咪唑菌酮(纯度≥96.9%)、呋霜灵(纯度≥98.5%)、吡喃灵(纯度≥93.9%)、苯酮唑(纯度≥96.8%)、枯草隆(纯度≥97.6%)、敌草隆(纯度≥99.3%)、乙环唑(纯度≥97.2%)、扑灭津(纯度≥94.8%)、丙环唑(纯度≥99.0%)、西草净(纯度≥98.9%)、噻苯隆(纯度≥99.0%)、抗霉素(纯度≥99.0%)、杀虫脒(纯度≥97.1%)、苯氰菊酯(纯度≥99.1%)、硫敌草(纯度≥98.1%)、倍硫磷(纯度≥98.0%)、氧倍硫磷(纯度≥95.7%)、倍硫磷砜(纯度≥98.0%)、倍硫磷亚砜(纯度≥99.5%)、氯氟胺氰戊菊酯(纯度≥98.4%)、异稻瘟净(纯度≥97.5%)、氧甲拌磷(纯度≥95.7%)、氧甲拌磷砜(纯度≥93.6%)、杀虫畏(纯度≥98.7%)、灭鼠灵(纯度≥99.4%)、喹禾灵(纯度≥94.5%)、氟氯氰菊酯(纯度≥99.70%)、氯氰菊酯(纯度≥99.52%)、氰戊菊酯(纯度≥87.0%)、禾草灵(纯度≥99.8%)、除线磷(纯度≥96.7%)、稗草畏(纯度≥92.1%)、灭多威(纯度≥99.0%)、保棉磷(纯度≥99.0%)、苯线磷(纯度≥99.0%)、涕灭威(纯度≥99.0%)、涕灭威砜(纯度≥99.2%)、涕灭威亚砜(纯度≥99.0%)、氟虫腈(浓度100 μg/mL)、氟甲腈(纯度≥99.5%)、氟虫腈硫化物(纯度≥99.1%)、乙硫磷(纯度≥99.0%)、甲氰菊酯(纯度≥99.0%)、甲基对硫磷(纯度≥99.0%)、氧化乐果(纯度≥99.1%)、敌敌畏(纯度≥99.2%)、马拉硫磷(纯度≥99.3%)、甲拌磷(纯度≥98.9%)、杀扑磷(纯度≥99.6%)、久效磷(纯度≥99.0%)、稻丰散(纯度≥99.0%)、三唑磷(纯度≥99.2%)和甲基乙拌磷(纯度≥99.1%),以上标准品购于德国 Dr.Ehrenstorfer-Schafers公司;二乙酰镳草镰刀菌烯醇(纯度≥95.0%)、新茄病镰刀菌烯醇(纯度≥95.0%)、雪腐镰刀菌烯醇(纯度≥95.0%)、镰刀菌烯酮(纯度≥99.9%),以上标准品购于北京曼哈格生物科技有限公司;三唑酮(纯度≥98.7%)、三唑醇(纯度≥98.8%)、灭多威(纯度≥99.9%),以上标准品购于德国LGC公司;阿维菌素(纯度≥96.5%)、腈菌唑(纯度 100%),以上标准品购于德国WTEGA公司;甲基毒死蜱(纯度≥99.9%)、丙溴磷(纯度≥98.2%)、黄曲霉毒素B1(纯度≥99.6%)、黄曲霉毒素 B2(纯度≥99.0%)、黄曲霉毒素 G1(纯度≥99.0%)、黄曲霉毒素 G2(纯度≥99.0%)、黄曲霉毒素 M1(纯度≥99.9%)、黄曲霉毒素 M2(纯度≥99.5%)、伏马毒素B1(纯度≥99.6%)、伏马毒素B2(纯度≥99.7%)、伏马毒素 B3(纯度≥99.0%)、赭曲霉素A(纯度≥99.2%)、赭曲霉素B(纯度≥99.5%)、赭曲霉素C(纯度≥99.3%)、展青霉素(纯度≥99.1%)、玉米烯酮(纯度≥99.3%)、杂色曲霉毒素(纯度≥99.6%),以上标准品购于美国Sigma公司;HT-2毒素(纯度≥99.0%)、T-2毒素(纯度≥99.0%)、3-乙酰脱氧瓜萎镰菌醇(纯度≥99.0%)、15-乙酰脱氧瓜萎镰菌醇(纯度≥99.0%),以上标准品购于青岛普瑞邦生物工程有限公司;脱氧雪腐镰刀菌烯醇(纯度≥95.0%)购于美国O2si公司。
试剂:甲醇、乙腈、甲酸(色谱纯),美国Fisher公司;高效基质脂肪吸附柱(EMR-Lipid2),美国Agilent公司;甲酸铵(色谱纯),上海安谱公司。实验用超纯水为Milli-Q水处理系统制得。
四极杆/静电场轨道阱高分辨质谱Q-Exactive(美国ThermoFisher公司),配有HESI Ⅱ离子源,液相色谱系统为 Dione UltiMate 3000高压液相色谱;BECHMAN Coulter Avanti J-26XP超高速冷冻离心机;平行定量浓缩仪、再循环冷却系统 B-740、真空泵V-700/701(瑞士 BUCHI公司);涡旋震荡器(德国IKA);电子天平(DTY-B1200,福州华志科学仪器有限公司)。
1.2 标准溶液配制
准确称取上述标准品约5.0 mg,于各自的5 mL容量瓶中,用乙腈溶解并定容至刻度,制成质量浓度为1 mg/mL的标准储备液,置于4 ℃冰箱中保存。
混合标准工作液:准确吸取适量的储备液用乙腈配成各相应浓度为1 μg/mL混合标准工作液。

表1 140种化合物质量分析和色谱分析参数Table 1 Parameters of MS and HPLC for 140 compounds
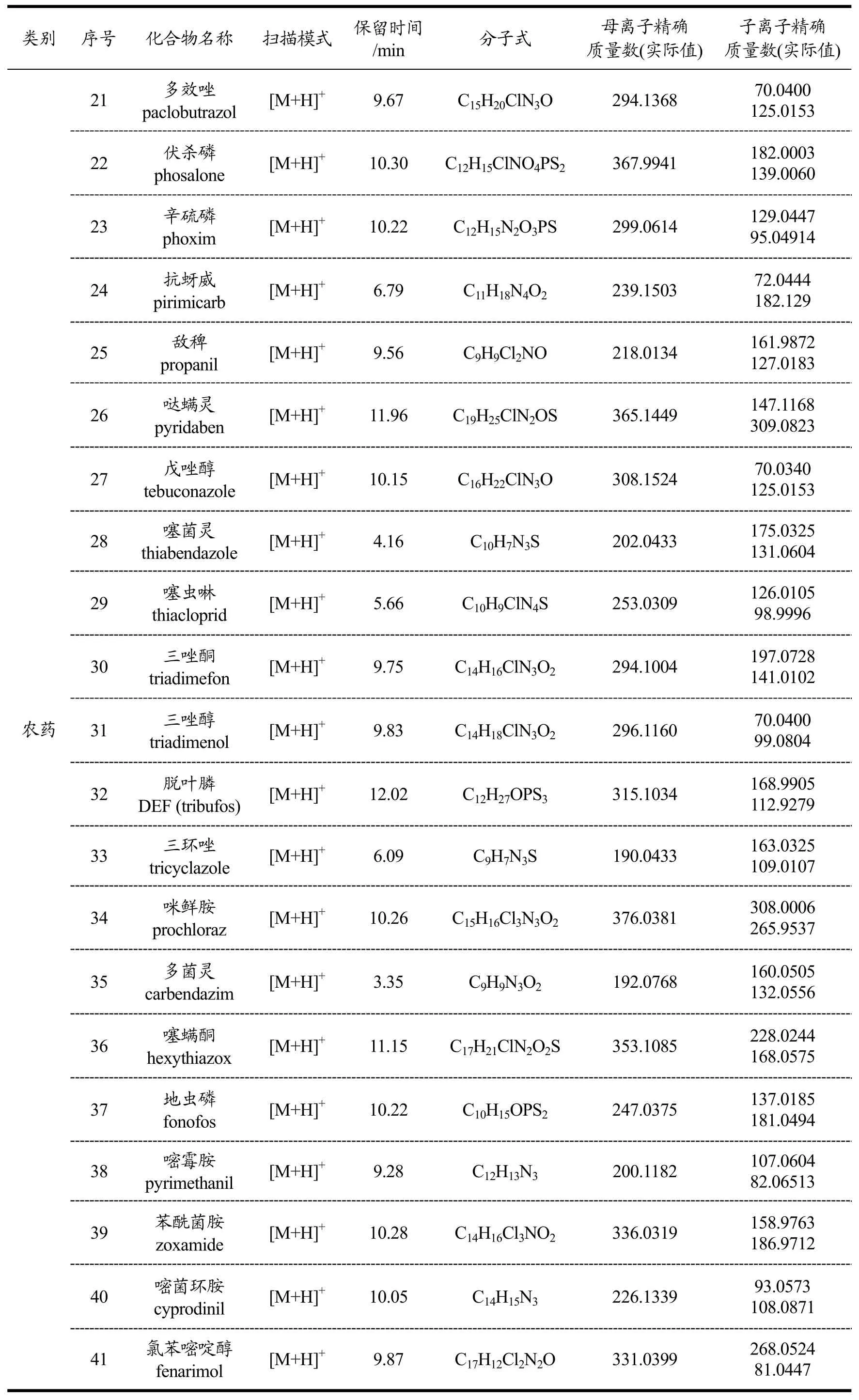
续表1
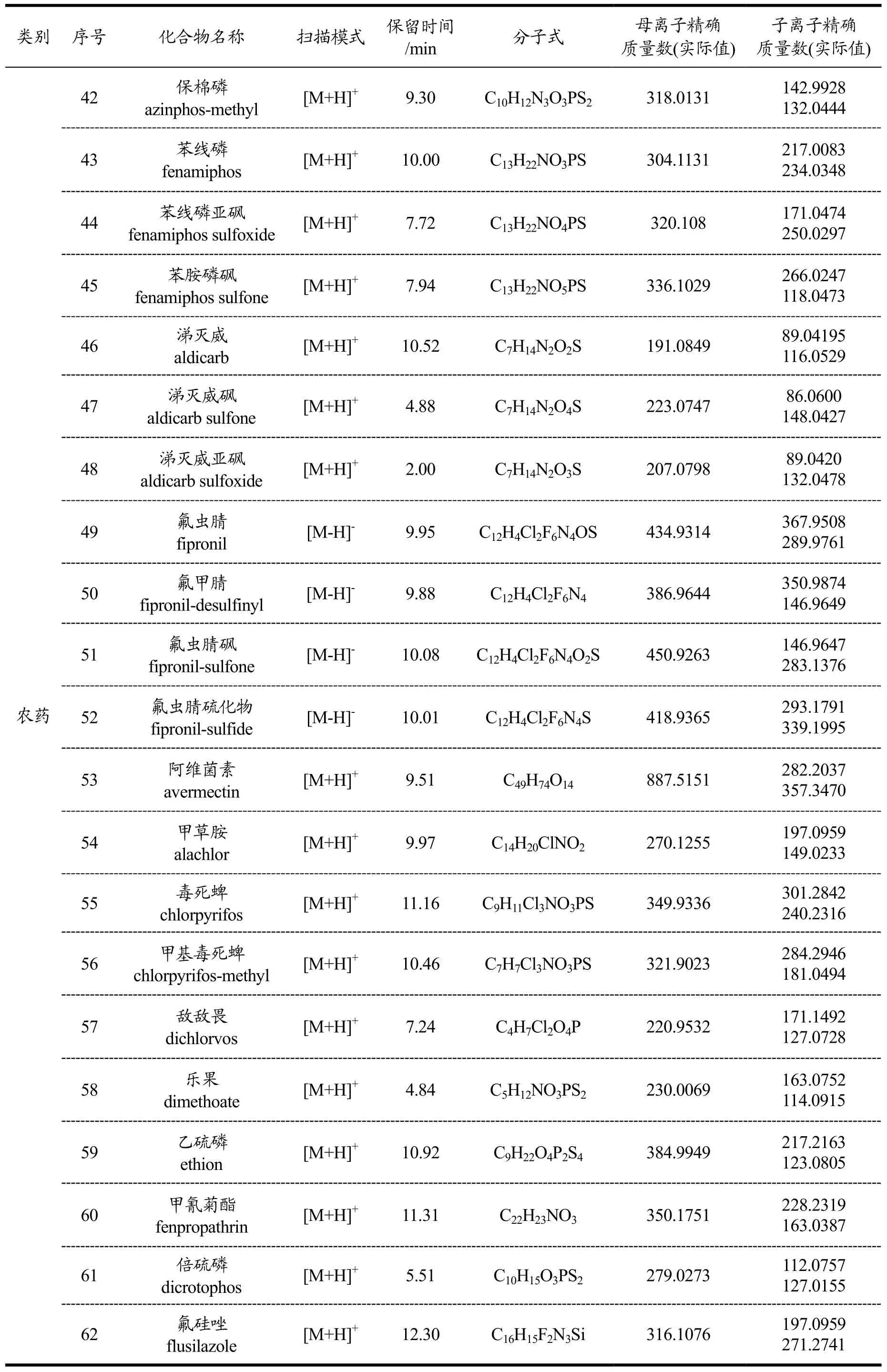
续表1

续表1

续表1
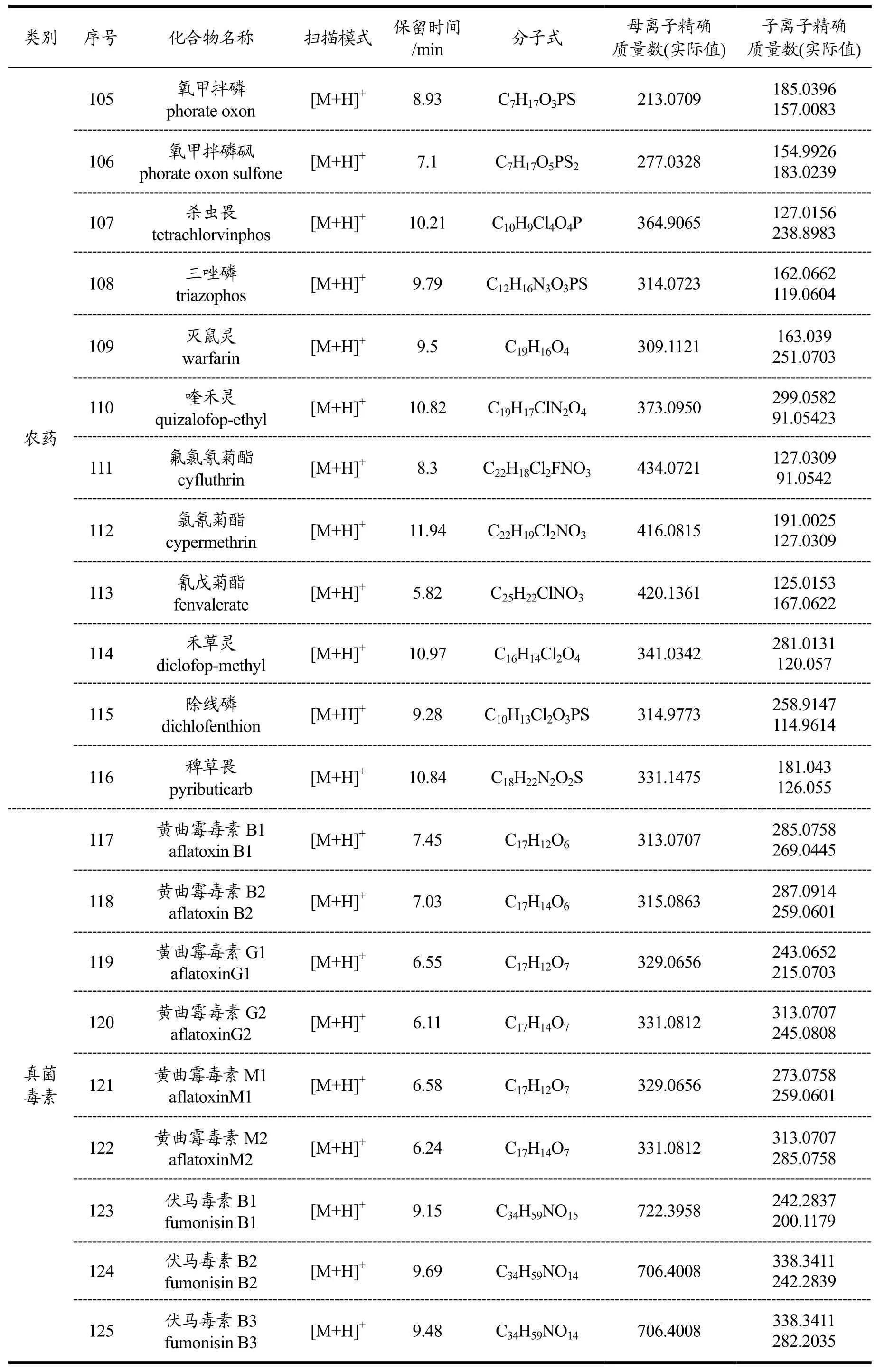
续表1

续表1
1.3 液相色谱条件
1.3.1 色谱条件
色谱柱:Waters Atiantis○RT3 C18 150 mm×2.1 mm,3.3 μm;柱温:40 ℃;进样量:10 μL;流动相:A为5 mmol甲酸水溶液(含0.1%甲酸铵),B为5 mmol甲酸甲醇(含0.1%甲酸铵)溶液。梯度洗脱程序0~2.0 min,保持25% B;2.0~7.0 min,流动相B的比例由25%线性变化至65%;7.0~8.0 min,流动相B的比例由65%线性变化至90%;8.0~12.0 min,保持90% B;12.1~15 min,保持25% B。
1.3.2 质谱条件
加热电喷雾离子(HESI)源温度为350 ℃;离子传输温度为320 ℃;鞘气为40 unit;辅助气为40 unit;毛细管电压为3.2 kV;离子传输管温度为325 ℃。Full scan/ddms2扫描模式:采集范围为80~1000 u,正负切换采集;一级质谱分辨率为70000 FWHM,二级质谱分辨率为17500 FWHM;碰撞池能量(NCE)为20、40、60 eV,见表1。
1.4 样品预处理
准确称取匀浆试样5.0 g(精确至0.01 g)于50 mL具塞离心管中,其中水产品制品试样加入10 mL乙腈/水(70:30,V/V)溶液,水产品试样加入10 mL乙腈/水(90:10,V/V)溶液,涡旋震荡 2 min,超声波提取10 min,4000 r/min离心10 min,上清液加入到300 mg EMR-Lipid柱中,自然流速,收集滤液以 15000 r/min,温度为5 ℃离心10 min,上层清移至15 mL离心管中于40 ℃平衡定量浓缩仪中浓缩至干。加1.0 mL甲醇/水(25:75,V/V)溶液涡旋溶解残留物,提取液过0.22 μm滤膜,供超高效液相色谱-高分辨质谱仪测定。
2 结果与讨论
2.1 色谱条件优化
研究选择对多组分分离较好的三款色谱柱Waters Atiantis○RT3 C18(150 mm×2.1 mm,3.3 μm)、Thermo Accucore RP-MS C18(100 mm×2.1 mm,2.6 μm)和ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm)对140种药物进行分离,结果显示Thermo Accucore RP-MS C18(100 mm×2.1 mm,2.6 μm)色谱柱对3-乙酰脱氧瓜萎镰菌醇和 15-乙酰脱氧瓜萎镰菌醇无法进行有效的分离;野燕枯、氟虫腈、氟甲腈、氟虫腈砜、氟虫腈硫化物在ACQUITY UPLC BEH C18(100 mm×2.1 mm,1.7 μm)质谱响应值较低;140种目标化合物在Waters Atiantis○RT3 C18(150 mm×2.1 mm,3.3 μm)形成的色谱峰峰形尖锐,对称性好,原因是色谱柱柱长对目标化合物的分离有较大影响,色谱柱越长分离效果越好、越稳定。
研究140种目标物中,4种目标化合物(氟虫腈、氟甲腈、氟虫腈砜和氟虫腈硫化物)在正离子模式下信号强度较弱,而在负离子模式下有较强的响应信号,因此为负离子模式检测。其他目标化合物均为正离子模式检测,为了提高正离子模式目标物的响应值,本研究在流动相(包括A和B)中均添加了甲酸,以便提高目标化合物的灵敏度。比较了0.1%甲酸水-0.1%甲酸乙腈、0.1%甲酸水-0.1%甲酸甲醇、5 mmol甲酸水溶液(含 0.1%甲酸铵)-5 mmol甲酸乙腈溶液(含0.1%甲酸铵)、5 mmol甲酸水溶液(含0.1%甲酸铵)-5 mmol甲酸甲醇溶液(含0.1%甲酸铵)对140种目标化合物的质谱响应和色谱分离的影响,结果发现使用5 mmol甲酸水溶液(含0.1%甲酸铵)-5 mmol甲酸甲醇溶液(含0.1%甲酸铵)为流动相,140种目标化合物的质谱响应较好,色谱峰峰形尖锐,对称性好,也能够达到检测要求,所以本研究选择5 mmol甲酸水溶液(含0.1%甲酸铵)-5 mmol甲酸甲醇溶液(含0.1%甲酸铵)为流动相(见图1)。
2.2 质谱条件优化
本方法以流动注射方式对 140种目标物在正负切换离子模式下进行一级全扫描,采用Q Exactive高分辨质谱的一级母离子全扫描加数据依赖的二级子离子扫描模式(Full MS/dd-MS2),设定涵盖目标物的质量数范围(m/z150~1000)进行一级全扫描,并以每个化合物的理论质量数(表1)建立二级扫描的目标列表。在实际扫描过程中,当一级全扫描发现目标列表里的母离子时,且信号强度超过预设值后,就会触发数据依赖子离子扫描模式,进而获得对应母离子精确质量数的二级离子全扫描质谱信息,以实现定性确证。根据欧盟 2002/657/EC对禁用药物残留检测确证方法的要求,确证检测需要 4个鉴别点。本实验通过上述质谱参数的优化和筛选,每种待测物最终确定 1个监测离子对,以满足检测确证的要求(见表1)。
2.3 前处理条件优化
2.3.1 萃取溶剂的优化
研究选择阴性干制鲈鱼样品为实验材料,添加浓度为5.0 μg/kg的混合标准品溶液,每个样品3平行,外标法定量对萃取溶剂(乙腈/水(70:30,V/V)溶液(含 1%甲酸)、甲醇/水(70:30,V/V)溶液和乙腈/水(70:30,V/V)溶液)进行优化。结果显示乙腈/水(70:30,V/V)溶液(含1%甲酸)和乙腈/水(70:30,V/V)溶液均能回收到140种药物残留,甲醇/水(70:30,V/V)溶液只能部分回收到目标化合物,尤其是炔草酯、炔草酸、霜脲氰、野燕枯、氟硅唑等药物几乎回收不到,乙腈/水(70:30,V/V)溶液(含 1%甲酸)虽然能够回收到140种化合物,但是HT-2毒素、伏马毒素(B1、B2和 B3)、新茄病镰刀菌烯醇和部分农药残留回收低于30%,使用乙腈/水(90:10,V/V)溶液作为萃取溶剂,140种化合物回收均高于70%,因此本研究选择作为乙腈/水(70:30,V/V)溶液萃取溶剂,比较三种萃取溶剂,回收率变化较大的40种目标化合物见图 2。鲜活水产品由于样品基质中有较多水分,经比较萃取溶剂乙腈/水(90:10,V/V)溶液、乙腈/水(80:20,V/V)溶液和乙腈/水(70:30,V/V)溶液,结果显示乙腈/水(90:10,V/V)溶液作为萃取溶剂既能保证140种目标化合物回收率大于70%,又能减少滤液中水分对目标化合物的干扰,减少了浓缩时间,提高了检测效率。
2.3.2 高效基质脂肪吸附剂的优化
高效基质脂肪吸附剂是一种能够选择性去除复杂基质中脂肪含量的独特吸附剂,具有快速、易操作和净化能力强等特点。本研究选择阴性干制鲈鱼样品为实验材料,添加浓度为5.0 μg/kg的混合标准品溶液,每个样品3平行,外标法定量对600 mg和300 mg两款EMR-Lipid柱进行比较分析,结果显示,相比300 mg EMR-Lipid柱,600 mg EMR-Lipid柱去除脂质效果更好,但是由于EMR-Lipid会对部分目标化合物产生较强的吸附作用,使用600 mg EMR-Lipid柱会导致部分目标化合物的回收率低于60%,研究最终确定300 mg EMR-Lipid柱为去除干扰物、保证灵敏度的最佳选择。
2.3.3 离心条件和浓缩条件的优化
虽然EMR-Lipid已经去除了样品中大部分油脂等干扰组分,但是样品提取液中还残留少量的油脂、蛋白等组分,研究采用高速冷冻离心对样品提取液进行处理,能够有效的促进油脂凝结和蛋白质的沉降,减少其对目标化合物检测的干扰。本研究对样品提取液进行了浓缩,选择平行样品定量浓缩仪减压浓缩样品提取液至近干,并对浓缩的条件进行摸索,确定浓缩温度为45 ℃;冷凝温度为-2 ℃;真空度采用梯度下降的方式(250 kPa 5 min,80 kPa 5 min,30 kPa浓缩至干)。该方法可以同时浓缩处理 24份样品提取液,完全浓缩至干仅需1 h。该处理方法能够有效的降低检测成本,提高检测质量和效率。
2.4 方法学验证
2.4.1 标准曲线、线性范围和检出限
在高分辨质谱的分析中,其提取的精确质量数色谱图无基线噪音,因此采用传统信噪比方法无法定义方法的检出限。本方法采用基质标准曲线的 y轴截距除以斜率的标准偏差的3.3倍以上数据确定检出限。准确量取适量混合标准储备工作液,用乙腈/水(10:90,V/V)溶液稀释成一系列浓度梯度的标准品工作液,在1.3节条件下依次测定。结果显示各种化合物在质量浓度范围内线性良好,相关系数(r)均大于0.991,检出限为 0.02~0.4 μg/kg(见表 2)。
2.4.2 方法准确度和精密度
为验证本方法的精密度,设计以下实验方案: 在阴性水产品样品(包括草鱼、罗氏虾、干制鲈鱼和干制鲍鱼)中进行加标回收试验,并做了精密度试验。在每一种水产品样品添加浓度为 2.0、4.0、20 μg/kg三个浓度水平6次平行加标实验中,结果显示,平均回收率在 70.1%~109.1%之间,相对标准偏差在1.0%~14.1%之间,说明本方法的准确度和精密度满足要求。
2.4.3 实际样品检测
利用本研究建立的分析方法检测 39批实际样品(其中包括5批鲈鱼样品、10批罗非鱼样品、5批干罗氏虾样品、8批干制鲈鱼样品、8批干制草鱼和 3批干制鲍鱼样品),其中在 1批干制草鱼中检出毒死蜱,检出量为4.32 μg/kg。
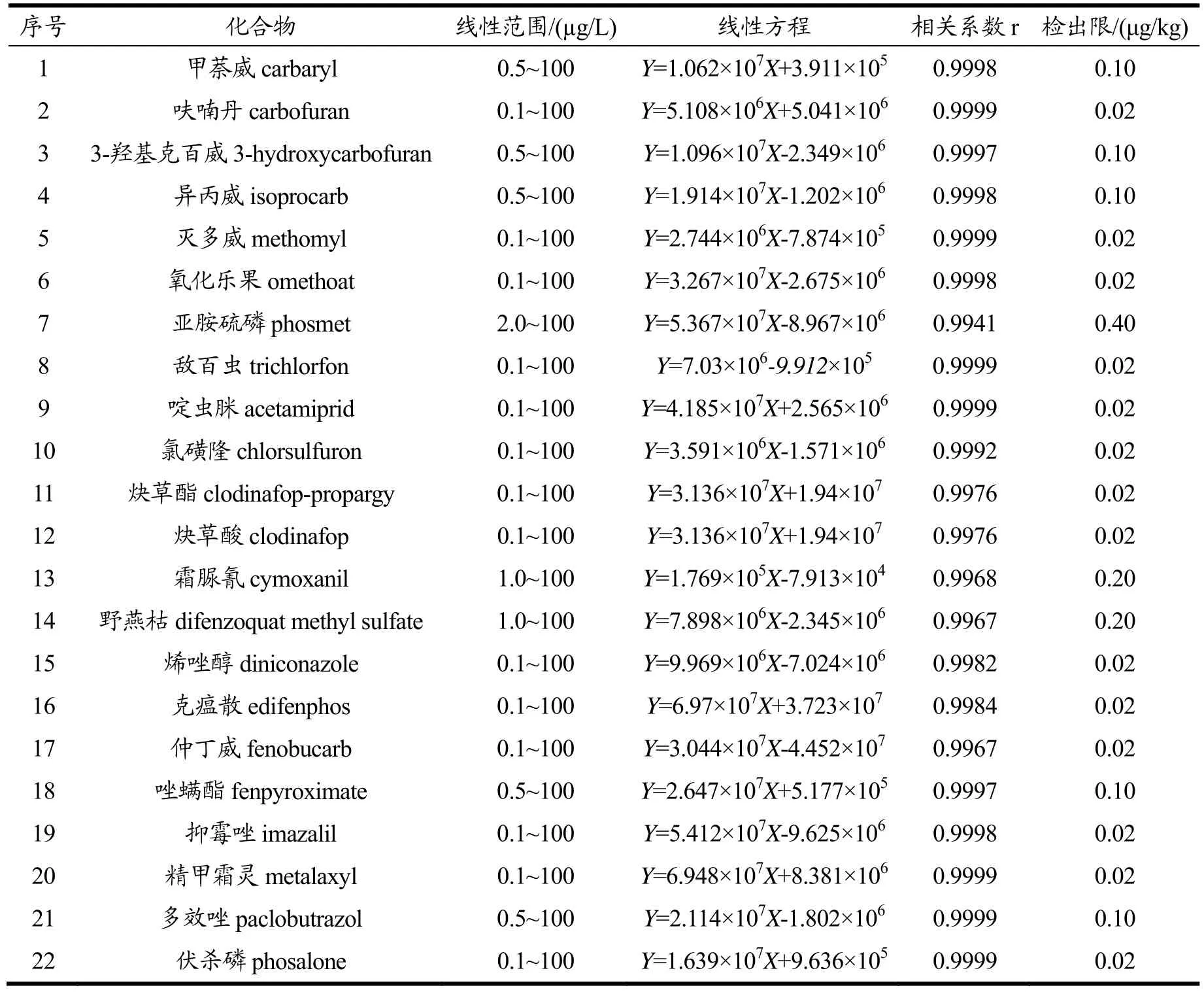
表2 140种目标物线性回归方程和相关系数Table 2 Regression equation, correlation coefficient of the 140 compounds

续表2

续表2

续表2
3 结论
本研究以水产品及干制水产品制品为实验材料,以乙腈水溶液为提取剂,前处理过程中采用一种新型脂肪吸附剂EMR-Lipid柱去除样品基质中脂肪和磷脂等杂质的干扰,随后用平行定量浓缩仪进行浓缩,建立了检测水产品及其制品中116种农药和24种生物毒素残留的超高效液相色谱-四级杆/静电场轨道阱质谱法(UPLC-Q Exactive Orbitrap MS),该方法可同时定性定量,具有灵敏、快速、简单、省时等特点,方法学结果满足GB/T 27417-2017《合格评定 化学分析方法确认和验证指南》的技术要求。
猜你喜欢
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
粉末冶金技术(2021年3期)2021-07-28
科学与财富(2021年13期)2021-07-04
生物工程学报(2020年6期)2020-07-31
食品界(2018年8期)2018-09-03
中学生数理化·高二版(2016年6期)2016-05-14
中小企业管理与科技·中旬刊(2014年7期)2014-09-24
同位素(2014年2期)2014-04-16
中国信息化·学术版(2013年3期)2013-06-25
