
多性状多位点遗传关联分析的统计方法研究及其应用进展
2022-01-28 07:46:54潘东东
广西师范大学学报(自然科学版) 2022年1期
艾 艳, 贾 楠, 王 媛, 郭 静, 潘东东
(云南大学数学与统计学院, 云南昆明650500)
人类发展进程中,对复杂疾病致病机理的研究一直是生物医学领域的热点和难点问题。目前,科学界已形成一个普遍共识,即大多数复杂疾病或性状是多对等位基因协同互作的结果。单个基因对疾病或性状的影响微效,而多个微效基因却可以通过加性效应影响性状表达或疾病发生。此外,这些微效基因还会与环境产生交互作用,其等位基因之间具有显性不完全性质[1-2]。
国际人类基因组计划之后的后基因时代,研究人员开始转向以基因组为对象分析生物遗传现象,而全基因组关联研究(genome wide association studies,GWAS)被公认为是最具前瞻性,且运用范围最为广泛的方法之一[3-4]。GWAS通过对研究对象的整个基因组的基因或位点进行分型,并充分运用生物统计学和生物信息学等科学技术手段,旨在检测出与疾病关联的单核苷酸多态性(single nucleotide polymorphism,SNP)位点,以探究疾病或性状的潜在机理。近10年来,GWAS已成功鉴定出大量与人类复杂疾病或性状(比如,动脉粥样硬化、克罗恩病和糖尿病等)相关联的遗传变异位点。研究发现常见变异对常见疾病和罕见疾病均有影响,例如:位于ARSA基因上的rs2071421及SORT1基因上的rs599839常见变异位点突变影响二型糖尿病和低密度脂蛋白,其突变频率分别为0.47和0.312 7;而通过EXOC4基因上的rs12538505变异位点对孤独症的影响可看出,常见变异与罕见疾病同样密不可分[5]。这些研究结果给复杂疾病或性状的深入研究提供了全新视角。
目前,研究人员已为GWAS开发了许多针对常见变异的统计分析方法,而针对低频或罕见变异的探查方法却十分有限,导致能被GWAS完全解释其遗传机理的复杂疾病或表型仍然很少。许多研究表明,罕见变异对复杂疾病或性状的发生具有决定性影响和作用,且罕见变异对常见疾病和罕见疾病均有影响。例如:成纤维细胞生长因子受体-3(FGFR3)的罕见变异位点突变及SETD1A上的一个缺失变异,导致软骨发育不全和阅读障碍症等罕见疾病;位于TREM2基因上的rs75932628及APOC3上的rs138326449位点突变,引起阿尔兹海默症和甘油三脂高等常见疾病,其位点突变频率分别为0.006 3和0.001[6]。随着下一代高通量测序技术的快速进展,研究人员已经鉴定出数以百万计的罕见遗传变异,使基因分析得到爆炸式发展。
然而,罕见变异的研究面临许多问题。从数据处理来看,罕见变异在人群中极为少见,故罕见变异的最小等位基因频率(minor allele frequency,MAF)的值通常定义在0.05%~0.5%,甚至低于0.05%,导致针对罕见变异的研究通常需要收集超大规模的样本(比如10 000人以上),相应研究设计与执行存在一定困难;从统计方法来看,现有针对常见变异的关联研究方法不适用于罕见变异的统计分析。因此,有必要探索一种高效、可靠、可同时分析常见变异和罕见变异的多性状多位点统计分析方法,从而能高效探查基因信息中潜在的高维和结构化性质,实现更为精准地探究复杂疾病或性状的发生机理,为进一步开展更高效的GWAS研究提供统计理论和方法支持。事实上,这也是当前遗传统计学和生物信息学领域研究人员迫切希望能取得突破的问题之一。接下来,本文将对近年来研究人员开发的多种针对复杂疾病或性状的全基因组关联研究方法进行系统性概述,为生物医学应用研究人员在统计分析方法选择上提供一定理论参考。
1 遗传关联分析的统计方法
关联分析是一种探究复杂性状或疾病与风险基因之间关联性的统计遗传方法,它通过探索病例组与对照组中基因功能和位点差异,找出基因与复杂性状或疾病之间的关联效应,并充分运用研究群体的信息来精确检测遗传致病位点,是探究复杂性状或疾病潜在机理的主流分析工具。关联分析通常分为候选基因关联分析和全基因组关联分析。本文主要对全基因组关联分析进行探讨,并从多性状分析、单位点分析和多位点分析3个方面对近年来研究人员提出的全基因组关联分析方法进行概述。
1.1 遗传关联分析的研究现状
2013年以来,随着人类基因组计划(the human genome project,HGP)和国际人类基因组单体型图计划(the international hapmap project,HapMap)取得突破性进展,以及下一代测序技术的广泛应用,研究人员已开发出数量众多的多位点关联分析方法,以探究影响人类复杂性状或疾病发生的常见变异及罕见变异。图1根据中国知网近8年来涉及到的“全基因组关联分析”发文量的统计数据绘制而成。

图1 知网涉及GWAS的发文量
1.2 遗传关联分析存在的问题与挑战
在GWAS中,研究人员通常会面临统计学方法适用上的严峻挑战,即研究的个体数相对较少,而遗传标记或SNP位点的数量超大,这违背了一般统计模型的假定条件,使参数估计及统计检验难以实现。另外,绝大多数全基因组关联分析方法的结论都是在假定表型服从正态分布下得到,若因试验误差等原因造成表型分布出现位移或偏态,则会使统计分析的结果产生偏差。此外,大多数现有关联分析方法或多或少存在对复杂疾病或性状的基因检测不足等问题。
注意到高效的遗传统计研究方法不仅要能够有效检测遗传变异的效应方向,还应考虑遗传变异关联研究时相关协变量(如性别、年龄等)的影响,并对其进行恰当校正。另外,样本容量越大越有利于减弱关联分析中不利因素的干扰,检验与疾病或性状相关的突变位点的能力越强。因此,在关联分析中,尤为强调足够样本容量的重要性;同时,群体分层等混杂因素以及位点间潜在的相关性,都可能会导致假阳性率增高,所以,在遗传关联检验中应该将群体分层和潜在的交互效应考虑进去,发展更为快速、高效的全基因组关联分析方法。
1.3 多性状分析
越来越多证据表明,1个遗传变异可能影响多个性状,特别是一些复杂的人类疾病。因此,在GWAS框架下,探究将多个相关性状的关联分析方法用于复杂性状风险基因定位,以揭示性状遗传模式和遗传作用机理具有重要的理论价值和现实意义。近10年来,研究人员已提出多种联合分析多个相关性状的统计方法,常用的有:Aschard等[7]提出的主成分分析方法;Ferreira等[8]提出的典范相关分析方法[8];Bottolo等[9]提出的多个依变数的线性回归分析方法、聚类线性组合(clustering linear combination,CLC)方法;Bolormas等[10]提出的基因型对表型或性状的反转回归(MultiPhen)方法;Xu等[11]提出的偏最小二乘法分析方法等。从目前文献引用可以看出,将多个相关性状联合分析的混合线性模型方法更易被应用工作者接受。大量研究表明,多性状关联分析较单性状有更高的统计功效和精度。
1.4 单位点分析
在GWAS中,检测遗传变异和复杂性状之间的标准方法是加性基因模型下的单位点分析策略,即利用某一种检验方法或标准,对每个SNP位点与性状之间的关联性分别进行检测,通常采用线性回归和逻辑回归,分别对数量性状和离散性状进行评估。近年来,针对离散性状和数量性状,研究人员已经开发了诸多单位点检测方法。
1.4.1 单位点分析方法
1.4.1.1 离散性状
针对离散性状数据(如疾病-对照数据),传统的罕见变异研究方法有基于等位基因频率的对比检验[12]、Cocharan-Armitage趋势检验[13]、卡方检验[14]、基于哈代-温伯格不平衡对比检验[15]、Fisher精确检验[16]以及逻辑回归检验等。其中,Cocharan-Armitage趋势检验和逻辑回归检验较为常用,前者将位点上风险等位基因数量的增加看做是有序的,后者通过检验原假设以确定性状的随机变量与基因的随机变量之间的关联大小。这些检验通常需要根据病例和对照样本中每个位点的3种基因型的赋值建立2×3列联表,之后对病例和对照样本中基因型的频率差异进行检验。假设在GWAS中进行基因型测序时共收集r个病例和s个对照,样本量为n,其中n=r+s。记病例组和对照组中基因型为(GG,Gg,gg)的人数分别为(r0,r1,r2)和(s0,s1,s2),可得到基因型数据汇总表(见表1)。

表1 病例和对照组中的基因型频数
1.4.1.2 数量性状
针对数量性状,通常处理方法是将每个SNP视为自变量,将表型值视为因变量,建立线性回归模型,并对模型中各SNP与性状之间关联显著性进行t检验,以获取SNP与数量性状关联检验的P值。在建立线性回归模型时,还可以加入性别、年龄等协变量,用于排除各种混杂因素的干扰,从而提升关联性检验的准确性。
基因连锁不平衡(linkage disequilibrium,LD)的存在以及基因长度的增加都会导致检验功效降低。已有学者提出多种改进方法,即通过获取这些位点上基因的单倍型信息来进行单倍型分析。例如,Schaid[17]提出一种修正的关联检验Max方法,先分别计算j个位点上检验统计量的值Tj,然后定义检验统计量为
(1)
该检验统计量的精确分布未知,故通过重抽样方法计算检验的P值。当只有一个易感位点时使用该方法功效显著,但当多个易感位点共同作用于疾病时,其检验功效会大幅降低。
1.4.2 单位点分析不足之处
单位点检验方法的发展和应用已经较为成熟,但此类方法也存在一些不足,例如:单位点分析方法忽略了位点之间的关联性,难以检测到中等偏低效应的位点;另外,对n个SNP位点进行n次独立关联检验,通常需要对显著性水平α进行校正,以控制群体错误发现率(family-wise error rate,FWER,即误判个数大于1的概率)。最常用的显著性水平校正方法是邦费罗尼校正(Bonferroni correction),即通过校正独立假设检验的数量n来控制总体假阳率,其对P值要求非常严格,对每个位点而言,都需要极小的P值才能通过检验,致使在降低总体第一类错误发生率的同时也会使统计检验过于保守,容易造成总体假阴性率过高。因此,多重比较问题常导致单位点分析严重损失功效。另外,越来越多研究结果表明,大部分人类复杂性状或疾病是由多个SNP位点共同作用而引起,故研究人员逐渐尝试在GWAS中使用多位点分析策略。
1.5 多位点分析
作为单位点分析的升级方案,多位点分析通常将位于同一基因内的变异位点归为一个集合,然后对该集合中的多个遗传变异位点的累积效应开展研究,分析该集合中的位点与表型之间的关联程度,旨在进一步提高检验效能,并提供额外生物学可解释性。针对单位点分析的不足,多位点分析可利用SNP位点间的LD信息提高遗传关联检验的统计功效。
1.5.1 常用变异集分析方法
当下主流的多位点分析方法其基本策略都是对某个基因组区域内多个标记位点进行联合分析来实现,各方法的区别在于整合多个位点变异信息的方式。最直接的方法是对数量性状值与多个SNP位点基因型以及相关协变量建立多元回归模型。注意到该方法所用的检验统计量自由度等于所纳入的SNP位点数,当感兴趣的SNP位点数量较多时,会因自由度过大而降低检验效能[18-19]。因此,已有研究人员提出整合区域内全部SNP位点遗传信息的检验方法[20],即首先评估每组研究对象的SNP位点相似度,并将成对遗传相似性与成对性状相似性进行比较。这类方法中应用最广泛的有负荷检验、方差成分检验、综合检验和EC检验[21-28]。
1.5.1.1 负荷检验
负荷检验先将多个遗传变异信息分解为单一遗传得分[21-24],通过检验该得分与某一性状之间的关联,计算集合中所有变异的最小等位基因(MAF)来归集基因型信息。假设βj=wjβ,其中β为所有变异位点效应的基准常数。原假设H0∶β=0的负荷检验统计量为
(2)

常用的负荷检验,如队列等位基因和检验(cohort allelic sums test,CAST),通常假设疾病风险的增加与某个罕见变异存在密切关联[22]。如果一个感兴趣区域(region of interest,ROI)无最小等位基因,则设遗传评分为0,否则为1。多元折叠(combined multivariate and collapsing,CMC)方法则对罕见变异进行折叠,在不同的MAF类别中利用HotelingT检验来评估变异的关联效应。Morris等[23]、Madsen等[24]提出的加权和检验(the weighted-sum test,WST)则采用Wilcoxon秩和检验,通过置换运算计算对应的P值。
上述方法都是按照诸如同一通路或位点相邻等相似性原则,先将ROI内的罕见变异聚集在一起,再对SNP集中的位点进行关联检验。当因果变异比例高,且ROI内因果变异效应大小和方向相同时,这些统计分析方法具有较高功效,这是因为负荷检验依赖于一个强假设,即集合中的所有罕见变异与性状具有因果效应,若该假设不成立将会造成巨大功效损失[25-28],甚至当只有一小部分因果变异存在时,其统计功效也会显著降低。此外,大多数负荷检验均需要运用置换检验来估计P值,计算量大,耗时长。CAST和CMC可通过软件包EPACTS(http:∥genome.sph.umich.edu/wiki/EPACTS)实现。
为克服负荷检验的局限性,研究人员开发了几种自适应改进方法,这些方法在无效变异存在时也有相当稳健的表现,并允许增加或减少性状变异程度。Han等[29]开发了一种自适应和检验(adaptive sum test,ASum),它首先估计边际模型中每个变异的影响方向,然后根据对应方向进行负荷检验,但需要通过数据置换来计算P值。回归系数估计(estimated regression coefficient,EREC)方法则通过聚合基因内多个变异位点的突变信息,采用适当回归模型,将遗传信息与疾病或表型联系起来[30],该框架涵盖了常见的研究设计(例如:病例对照、队列设计和家系研究等)和常见的表型(例如:二元、定量和发病年龄等),并允许考虑任意协变量(例如:环境因素、祖先变量等)。该方法的等位基因频率阈值可设为固定的或可变的,罕见变异组合对表型的影响方向也不固定。模拟研究表明,该方法的统计功效与计算效率均较高。需要注意的是,当MAF很小时,EREC估计不稳定;同时,EREC检验通过在估计的回归系数上添加一个小常数来获得稳健估计,这可能会降低该检验的效率。变异阈值(variable threshold,VT)是另一种自适应扩展方法,对罕见变异的负荷检验选择最佳频率阈值,通过置换方法估计检验的P值[31]。基于核的自适应聚类(kernel-based adaptive cluster,KBAC)方法采用基于核自适应加权方法进行罕见变异关联检验,在一个连贯的框架中将风险或非风险变异分类与关联检验结合起来,并考虑协变量以控制潜在混杂因素,包括年龄、性别和人群子结构等。研究结果显示,KBAC较其他方法表现出更高功效,特别是存在基因交互作用的情况下[32]。
适应性检验比原始负荷检验更稳健,主要是由于它对每个SNP位点的潜在遗传结构的假设要求更少。但是,大多数适应性检验需要采取置换方式估计P值大小,存在计算强度大的弊端。这些方法可通过软件包Rvtests、VAT及SCORE-Seq(https:∥genome.sph.umich.edu/wiki/Rvtests、http:∥varianttools.sourceforge.net/Association/HomePage、http:∥dlin.web.unc.edu/software/score-seq)实现。
1.5.1.2 方差成分检验
基于随机效应模型的方差成分检验,通过评估一组变异的遗传效应分布来进行关联检验。常用方法有序列核关联检验(sequence kernel association test,SKAT)、平方和检验(sum of squared score,SSU)和C-alpha检验,都是通过汇总单个变异的得分检验统计信息来提升统计检验功效[27,33-34]。SKAT是一种灵活而强大的回归方法,它可以检测ROI内遗传变异中的离散或连续性状之间的关联大小,同时可以考虑相关协变量,以有效解决群体分层问题。具体地,SKAT假设回归系数βj独立同分布,其均值E(βj)=0,方差var(βj)=wj2T,无变异集效应的原假设为H0∶β=0,对应的SKAT检验统计量为
(3)
QSKAT在原假设下渐近服从混合卡方分布,其P值可快速解析获得,不仅有效提升计算速度,还能考虑变异之间的LD信息。该方法不需做任何假定就可从数据中估计出关联效应大小和方向,且能有效控制第一类错误率。然而,SKAT忽略了除单个基因或ROI之外的其他基因以及相关遗传信息,当密集因果变异方向基本一致时,采用SKAT进行统计分析将严重降低效能。另一类常用多位点分析方法SSU是先在逻辑回归模型框架下计算多个位点的得分向量及相关协方差阵,然后对每一个位点计算得分与方差分量的比值,最后将全部位点的比值加总得到多位点检验统计量。实际上,SSU与使用线性核的SKAT等价,且SKAT在不考虑协变量时也可简化为C-alpha检验[33-34]。这些方法可由R包SKAT和MiST (https:∥hsph.harvard.edu/skat/、https:∥cran.r-project.org/web/packages/SKAT/、https:∥cran.r-project.org/web/packages/MiST/)实现。
对于离散性状,在样本量和MAF较小时,基于大样本的P值计算方法难以控制第一类错误率。Lee等[35]开发一种基于矩的方法,使用检验统计量小样本精确方差和峰度估计调整零假设下的渐近分布。但是,当MAF非常低时这种调整可能不够,仍然需要置换方法获得较准确的P值估计。
1.5.1.3 综合检验
当ROI内有许多非因果变异或因果变异有不同关联方向时,方差成分检验比负荷检验更有效。相反,如果ROI内具有相同关联方向的因果变异占比高,则负荷检验比方差成分检验更有效。因此,已有学者提出将负荷检验和方差成分检验组合起来进行综合检验,以克服这2类方法的不足。常见的综合检验有SKAT-O、Fisher方法和MiST方法等。Derkach等[36]提出使用Fisher方法将2种检验的P值结合起来评估检验的显著性,其Fisher检验统计量为
F=-2ln(PSKAT)-2ln(PBurden) 。
(4)
式中PSKAT和PBurden分别为SKAT和负荷检验的P值。注意到构成Fisher检验统计量的2个原始检验是基于同一组数据,不可避免存在较强相关性,这就可能导致相当大的功效损失,特别是当其中一个检验的适用条件基本成立时。另外,在实际中,SKAT和负荷检验的P值计算工作量都很大,已有学者开发了一种能自适应地组合SKAT和负荷检验统计数据的综合检验(SKAT-O),它采用一种自适应过程来实现最优值的估计与最小P值的计算[36]。
1.5.1.4 EC检验
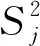
(5)
1.5.2快速检验方法
随着测序技术的飞速进步、测序成本的不断下降,数以千计的罕见SNP位点被纳入遗传关联研究中。注意到,对大量罕见SNP位点的统计计算属于密集型任务,为缩短计算机运算时间至合理范围,非常有必要寻求新的快速检验方法。接下来主要针对基于正则惩罚项和特征降维、贝叶斯理论、混合模型以及P值组合方法,回顾近年来开发的旨在提高计算速度、降低计算成本的若干快速检验方法。
1.5.2.1 基于正则惩罚项和特征降维的方法
在进行多位点关联分析时,特别是针对罕见变异,其样本数据大多具有维度高且样本量少的特点,即SNP位点数远大于样本量n。对此,Hoerl等[38]提出岭回归(ridge regression)分析法,该方法通过增加二次正则惩罚项来平滑模型系数,使线性模型在处理高维稀疏数据时更加稳定,可防止过拟合。注意,其系数压缩值是一个趋于0的近似值,故不能很好地解决共线性问题,导致模型解释性弱。Tibshirani[39]提出的Lasso (least absolute shrinkage and selection operator)方法,基于岭回归将L2范数形式的正则项改为L1范数,将待估参数中不重要的系数收缩至0,以获取特征层面的稀疏解。Lasso已被证明是一种有价值的模型拟合和特征提取工具。然而,Lasso倾向于从一组相关SNP位点中选择一个位点,而不能将它们全部选出,可能导致估计出的模型参数过于稀疏。
Zhang等[40]基于岭回归提出了一种可以处理模型过饱和的惩罚似然3该方法精确定位特定标记之间的相互作用,允许数量性状基因座(quantitative trait locus,QTL)效应缩小到零,而效应大的QTL几乎全部保留。惩罚似然方法将模型优化目标定义为参数先验分布与似然函数的结合,不需要考虑模型选择及相关问题,如模型的参数空间探索不足和计算密集问题等。研究表明,该方法可以处理比样本量大15倍的多个效应模型,可有效提升收敛速度。但该方法用于实际数据分析前常常需要进行一些修改,例如,标记可能不是精确沿基因型均匀分布的,尽管该方法不依赖于标记分布的一致性,但高度多重共线性、非常紧密的标记可能导致标记效应估计较差等不良影响。
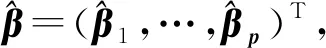

(6)

(7)
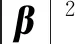
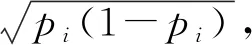
1.5.2.2 基于贝叶斯理论的方法
许多学者已将贝叶斯理论应用于多位点关联研究。Iwata等[43]提出基于贝叶斯关联分析的QTL检验方法,基本思想是在所有种群中对遗传效应使用相同参数进行建模,其统计模型为
(8)
式中:yi为性状值;qij为群体结构Q矩阵第i行第j列元素;αj是与种群j(j=1,2,…,J)相关的群体效应;xik表示第i个样本第k个标记的基因型值;γk是指示变量,假定每个QTL都在标记上,若第k个标记存在效应为βk的QTL,则γk=1,否则γk=0;εi服从正态分布。该模型考虑了多重QTL和群体结构的影响,运用阈值模型将性状观测值转换为潜在的连续变量,并通过Markov链、Monte Carlo方法同时映射多个QTL,得到对应参数估计值,可较好地控制单个QTL的假阳率和假阴率以及遗传效应估计误差。当只考察数百个标记时,该模型具有一定实用优势,但当标记数量大于2 000时,该方法收敛速度较慢;另外,由于标记等位基因与QTL等位基因之间连锁不平衡的模式和程度在群体之间各有不同,导致估计效应会存在差异[43]。对此,Park等[44]提出在经典贝叶斯分析的框架基础上添加效应惩罚,通过在其参数和方差上分别分配尺度混合正态(scale mixture of normal,SMN)先验和独立指数先验,得到双指数分布(Laplace)先验的贝叶斯Lasso方法,其分层模型结构可同时采用贝叶斯方法和似然方法来选择Lasso参数。为简化结果,贝叶斯Lasso引入多种先验形式以实现不同类型的惩罚,因此,贝叶斯模型已被广泛应用于GWAS中,但其在大样本情况下的计算速度尤其慢[45-47]。

(9)
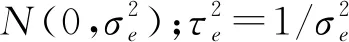
1.5.2.3 基于混合模型的分析方法
此外,有学者提出基于混合线性模型(mixed linear model,MLM)的分析方法来提高计算效率。MLM方法能有效控制种群结构和亲缘关系,但在处理超大型数据集时面临计算挑战。对此,Zhang等[49]提出的压缩混合线性模型(compressed mixed linear model,CMLM)方法将QTL效应视为固定效应,将样本聚类到若干个组中,降低有效样本大小,发展了预先确定总体参数(population parameters previously determined,P3D)算法。该方法通过简单使用原模型的预估方差分量,可避免检验时对其重复估计。这2种策略的联合使用显著缩短计算时间,并使统计分析功效得以维持或提高。为进一步提高CMLM方法的统计功效,Li等[50]提出优化压缩混合线性模型(enriched compressed mixed linear model,ECMLM)方法。ECMLM方法在CMLM方法的基础上增加1个新参数,使用3组亲缘关系算法和8种聚类算法之间的24种最佳组合,通过8种聚类算法将全部压缩级别的样本聚类为组。ECMLM方法可找到压缩级别、亲属关系和聚类算法的最佳组合,有效提升平衡统计功效和计算速度,且在某些情况下使用ECMLM代替CMLM所获得的功效增益比使用CMLM代替MLM所得到的改进更大。ECMLM方法可由R包GAPIT(https:∥maizegenetics.net/gapit)实现。
Kang等[51]提出对种群结构和遗传亲缘关系进行校正的有效混合模型关联(efficient mixed-model association,EMMA)方法。该方法将混合模型应用于关联映射,只需估计多个基因和误差的方差成分,从而计算速度和结果可靠性得到大幅提升。实际上,该方法是在Yu等[52]提出的模型Y=Xb+Zu+Wv+ε的基础上,将QTL效应u视为固定效应,并记β=(b,u),得到的新模型
Y=Xβ+Wv+ε,
(10)

在前人研究基础上,唐明生等[6]提出基于似然比检验的多位点关联分析方法,他们假设SNP位点效应服从正态分布,构建随机效应方差的LRT及限制性似然比检验(restricted likelihood ratio test,ReLRT)对多位点进行关联分析,并运用置换及Bootstrap方法获得统计量的零分布。然而,为获取更高精度P值而采用的重抽样方法计算尤为耗时,不适合大规模多位点关联分析。因此,Wang等[56]提出用混合分布来近似重抽样分布,以显著降低似然比检验的计算负担。该方法能有效控制第I类错误率,在保持良好统计功效的同时极大降低计算成本。
虽然某些情况下近似方法提供的结果与精确方法几乎一致,两者均可有效缩短计算运行时间。然而,在实践中,若没有进行精确计算,则很难判断近似方法的精确性,近似不准确必然导致功效降低。这方面尚待研究者进一步开发更为快速精确的基于混合模型的关联研究方法[57-61]。
1.5.2.4 基于P值组合的方法
近年来,研究者对结合样本P值或检验统计量的方法产生较大兴趣,开发了若干基于P值组合的方法,如:Tippett等[62]提出的最小P值检验;Donoho等[63]提出的针对性改进方案;Berk等[64]提出的Berk-Jones检验等。这些方法均是通过结合单个P值来聚合多个遗传效应。然而,在分析海量基因数据时,这些方法均存在计算负担大的缺点,特别是当一个P值组合检验需要执行多次,或当检验的P值均很小且个数极大时,采用置换法来求组合检验的水平不再可行。对此,Liu等[65]提出一类聚合柯西关联检验(aggregated Cauchy association test,ACAT)方法,实质上是一种通用而灵活的P值组合方法,其特别之处是只需要P值(和权重)作为输入,即可以根据柯西分布的累积密度函数得到ACAT的P值近似值,不需要考虑个别检验P值的相关性,其在零分布下的检验统计量为
(11)
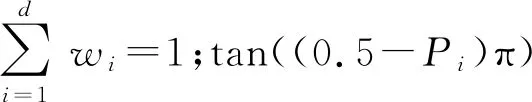
需要注意的是,当SNP位点的最小等位基因频率减小较多时(低于0.5%),使用ACAT方法的可靠度将无法保证。此时,若直接使用ACAT聚合某个区域内位点水平的P值,将导致过于保守的第一类错误率及过低的检验功效。为解决这个问题,研究人员已提出首先使用负荷检验来聚合最小等位基因计数(MAC)小于某个特定数量的变异,接着使用ACAT将罕见变异负荷检验的P值与感兴趣区域内其他变异的P值相结合[66]。该方法在区域内仅有少量几个因果变异时尤为有效,而ACAT-V在多个弱关联变异存在的情况下也会严重丧失功效[67]。ACAT方法可由R包ACAT(https:∥github.com/yaowuliu/ACAT、https:∥content.sph.harvard.edu/xlin/software.html)实现。
上述遗传变异统计分析方法初步解决了大规模SNP关联分析的部分计算问题。事实上,在遗传关联研究中,研究人员均期望在解决数据维度灾难的同时,能够用最少时间完成尽可能多的遗传变异位点检测任务。注意到现有方法对多种复杂疾病或性状与多个位点之间遗传关联度的探查效能不高,计算效率低,人们还需进一步发展更加有效的统计分析方法。科学合理地联合不同检验方法是可行策略,已有许多学者提出了多种两阶段分析方法,即分步骤对遗传变异进行系统分析,有效提高统计分析功效和计算速度[5,68-77]。
2 展望
现有联合检验方法大都具有稳健性和计算高效性,已有研究显示一些联合检验方法并不总是能提高检验功效[35,78]。目前已有的遗传变异关联研究方法中并不存在某一个检验在任何情况下都具有最优统计功效。
多位学者宣称其所提出的两阶段分析方法相比已有方法功效更高,但已有方法中绝大部分是针对单性状与多位点之间的关联性进行分析,小部分文献针对多性状与单位点进行探究。有理由相信,联合分析多性状与多位点之间的遗传效应可能会给疾病机理提供新的研究视角,但目前尚无公开发表的文献讨论相关统计方法及其应用,这有待研究人员进一步探讨和研究。
其次,当前大多数遗传关联分析策略都是针对常见变异与罕见变异二者之一来进行,然而,大量研究显示,遗传致病位点的等位基因频率分布非常广泛,很多复杂疾病或性状实际上是常见变异和罕见变异共同作用的结果。如果仅局限于研究单一类型的变异,可能会导致统计功效严重降低,无法探查出疾病或性状的真正机理。因此,联合分析常见变异和罕见变异以及它们之间可能的交互作用是必要的,目前相关文献有限,这是一个值得关注的研究方向。
再次,目前尚无一类公认绝对高效可靠的统计方法用于合并一组罕见变异进行联合分析,从探究最优位点合并方法的角度出发,寻求可检验多个罕见变异与多个性状之间关联性强弱且具有更优功效表现的统计方法是又一新的研究视角[26-27,30]。
最后,在单位点单性状的两阶段分析方法基础上,应考虑对多位点多性状遗传关联分析进行两阶段分析方法研究。
猜你喜欢
区域治理(2022年40期)2022-11-27 04:01:54
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
动漫界·幼教365(小班)(2019年10期)2019-10-28 02:04:20
动漫界·幼教365(大班)(2019年10期)2019-10-28 01:54:09
动漫界·幼教365(中班)(2019年10期)2019-10-28 01:53:17
当代陕西(2019年15期)2019-09-02 01:52:00
学苑创造·A版(2018年11期)2018-02-01 06:29:20
读者(2017年5期)2017-02-15 18:04:18
百科知识(2015年18期)2015-09-10 07:22:44
