
Tuning main-group s-block metal Mg as a promising single-atom electrocatalyst for N2 fixation: A DFT study
2022-01-24 14:06YANGKangWANGChanglaiDENGXiCHENQianwang
中国科学技术大学学报 2021年11期
YANG Kang, WANG Changlai,3, DENG Xi, CHEN Qianwang,2*
1. Hefei National Laboratory for Physical Sciences at the Microscale, Department of Materials Science and Engineering, University of Science and Technology of China, Hefei 230026, China;2. The Anhui High Magnetic Field Laboratory, Hefei Institutes of Physical Science, Chinese Academy of Sciences, Hefei 230031, China;3. Department of Materials Science and Engineering, Center of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Hong Kong 999077, China;4. School of Chemistry and Materials Science, University of Science and Technology of China, Hefei 230026, China
Abstract: The electrocatalytic nitrogen reduction reaction (NRR) can transform nitrogen and protons from aqueous electrolytes to ammonia by using renewable electricity under ambient conditions, which is a promising technology to replace the Haber-Bosch process. However, this technology is extremely challenging as it requires highly active electrocatalysts to break the stable triple-bonds of N2.With p bands,main-group s-block metals have been rarely explored in NRR compared with transition metals.Herein, we employ first-principles calculations to propose a Mg single atom catalyst as a promising high-performance electrocatalyst for NRR, where Mg atom is coordinated with four oxygen atoms within graphene (Mg-O4). Our results reveal that N2 can be efficiently activated on Mg-O4 and reduced into NH3 through the alternating mechanism. Moreover, ab initio molecular dynamics simulations demonstrate the Mg-O4 structure has high stability.
Keywords: electrocatalytic N2 reduction reaction; main-group metal; single atom catalyst; first-principles calculations
1 Introduction
Ammonia (NH3) is an essential headstone in global industrial chemicals as it is a fundamental raw feed to produce nitric acid, ammonium salt, and chemical fertilizers[1-3]. Currently, more than 90% of NH3is produced by the Haber-Bosch process[4]. However, this process requires high pressure (15-40 MPa) and temperature (350-550 °C), which consumes 1%-2% of the world’s annual energy supply[5-8]. Therefore, more attention has been paid to a greener and simpler way to product NH3. A promising way for the sustainable production of NH3under thrambient condition is the electrocatalytic N2reduction reaction (NRR), which could provide proton source via electrolysis of water molecules by renewable energy technology[9-12].However, the electrocatalytic NRR, involving cleavage of the strong dipole moment of the N≡N triple bond, is extremely challenging[13, 14]. Therefore, exploring highly active electrocatalysts for NRR is highly desirable.
Benefiting from the occupied and unoccupied d orbitals, transition metals have been widely applied for the electrocatalytic NRR. For example, a Fe-doped W18O49nanowire had been reported for the electrocatalytic NRR, which exhibited a high NH3Faradaic efficiency of 20.0% at an overpotential of -0.15 V versus reversible hydrogen electrode (RHE)[15]. In another case, a S-rich MoS2nanosheet achieved a high ammonia production yield rate of 43.4 μg per h per mg catalyst[16]. In contrast,main-group metals in s-block have been rarely explored in the electrocatalytic NRR.Nonetheless, in nature, metalloenzymes containing s-block main-group metals can catalyze many challenging biological reactions[17].For example, chlorophyll, consisting of a porphyrin ring with a central atom of magnesium, can transform CO2and H2O to carbohydrates by capturing the sunlight[18]. Calcium cofactor plays an important role in the ferroportin transport activity[19]. However, the poor electrical conductivity of metalloenzymes limits their application in the electrocatalytic NRR. Anchoring s-block main-group metal atoms onto an appropriate substrate with good conductivity may be a solution.
Graphene, consisting of a single layer of sp2-hybridized carbon, has high electric conductivity and stability, which is an ideal support to anchor metal atoms[20, 21]. For example, Yao et al. prepared atomically dispersed Ni on the defective graphene[22].Monodisperse cobalt atoms embedded in nitrogen-doped graphene was successfully synthesized by Ji’s group[23]. It is worth noting that the nitrogen atoms in catalyst might disturb its catalytic performance for NH3synthesis by electrocatalysis method. Hu and co-workers reported that tetragonal Mo2N would undergoes decomposition and result in the generation of ammonia, instead of catalyzing electrochemical reduction of N2[24]. Besides, synthetic steps for introducing nitrogen into graphene including calcination in NH3atmosphere or the use of compounds containing NH2components such as melamine and urea will also result in the false performance of catalysts[25, 26].
Motivated by these considerations, herein, we perform first-principles calculations to explore the potential of s-block main-group element Mg atom decorated by the defective graphene substrate for NRR under ambient conditions. Our results reveal that the structure of Mg atom anchored on the graphene substrate coordinating with four O atoms (Mg-O4) has a strong activation effect on nitrogen molecules, which could efficiently reduce N2into NH3by the alternating mechanism. Furthermore, the result of ab initio molecular dynamics simulations also demonstrated the thermal stability of Mg-O4at the room temperature.
2 Computational details
In this work, we implemented the Vienna Ab initio Simulation Package (VASP) to conduct all calculations through spin-polarized density functional theory (DFT)[27]. The exchange-correlation energy functional was described in the Perdew-Burke-Ernzerhof (PBE) generalized gradient approximation (GGA) with projector augmented wave (PAW) pseudopotentials[28, 29]. The cut-off energy of 400 eV were employed. The convergence threshold of energy and force were 1 × 10-5eV and 0.02 eV·Å-1, respectively. Thek-point grids are 5 × 5 × 1 for geometry optimization. In order to minimize the periodic interaction, a vacuum layer of 20 Å along the vertical direction was set. Chemical-bonding analyses were carried out with the computer program LOBSTER (Local Orbitals Basis Suite Towards Electronic-Structure Reconstruction)[30, 31]. Ab initio molecular dynamics simulations (AIMD) were employed to evaluate the thermodynamic.The time step is set as 2 fs and the simulation is performed using the canonical ensemble (NVT) in the temperature of 300 K with an optimized initial structure.
The binding energy (Eb) between the Mg atom and the O-modified graphene substrate was calculated by the following equation:
Eb=Ecatalyst-EO/C-EMg
Furthermore, the Gibbs free energy (ΔG) for each reaction step in different NRR pathways was defined as follows:
ΔG=ΔE+ΔEZPE-TΔS+ΔGU+ΔGpH
ΔEis the electronic energy difference between the product and the reactant species,ΔSandΔEZPEare the changes in entropy and zero-point energy, respectively.ΔGUrepresents the effect of applied bias and is equal to eU.ΔGpHis the contribution of H+and is equal to -kBT×ln10×pH, wherekBis the Boltzmann constant. The value of pH is set as 0 to produce thermodynamic values comparable to those of other works.
3 Results and discussion
3.1 Anchoring Mg atom by defective graphene
Previous studies have demonstrated that divacancy is the most common defect present in 2D graphene and it possesses impressive ability for anchoring transition metal atoms which present effective catalytic activity for some electrocatalysis reactions, such as O2reduction reaction (ORR), CO2reduction reaction[32-34]. The different coordination element around metal atom would result in diverse electron structure of metal active site. Thus, we design six models where Mg atom is coordinated with different number of O and C atoms. According to the number of O and C coordination atoms, we named those six models as Mg-C4, Mg-C3O1, Mg-C2O2, Mg-O2C2, Mg-C3O1, Mg-O4, respectively. The models of Mg-C2O2and Mg-O2C2represent the different coordination structure with Mg atom. The optimized structures of the six models are shown in Figure 1. There is no obvious distortion or collapse of O-modified graphene substrate. Moreover, when the number of O as coordination atoms is greater than two, the Mg atom is well anchored in the graphene layer as shown in the side-view of models. This indicates that the divacancy defect in graphene is also appropriate for anchoring the main group element Mg. Furthermore, the calculated binding energies range from -0.57 to -4.30eV (Figure S1), demonstrating that they are thermodynamically stable.

Figure 1. the optimized structure of six models for Mg-O/C. (a) Mg-C4, (b) Mg-C3O1, (c) Mg-C2O2, (d) Mg-O2C2, (e) Mg-C3O1, (f) Mg-O4.

Figure 2. The difference charge density of Mg-O/C with the adsorption of N2, cyan and yellow regions respectively represent positive and negative charge regions and the isosurfaces value is 0.006 e/Å3.
3.2 Adsorption of N2
It’s well-known that the chemisorption of N2molecule on the surface of catalysts is the prerequisite for an efficient NRR process. The effective chemisorption determines whether a catalyst is able to activate N2molecule. Previous studies have shown that side-on and end-on patterns are the two ways of N2absorption on the catalyst surface[35, 36]. We firstly verified both absorption modes of N2molecule on Mg atom. As shown in Figure S2, side-on absorption pattern changed into end-on mode after structure optimization in the all models. This result is in consensus with M. A. Duncan’s research conclusion, which demonstrated the activation of N2on Mg atom via the linear configuration rather than the side-on configuration[37]. Currently, it is widely believed that the activation of N2is via an “acceptance-donation” process[38], this mechanism indicates the mutual transfer of electrons between catalysts and N2molecule. Figure 2 presents the difference charge density of Mg-C/O with the adsorption of N2, where the charge accumulation and depletion can be clearly observed for both N2molecule and Mg-C/O catalyst. Mutual charge transfer in other five models indicates N2molecule is effective adsorbed by Mg atom anchored in the O-modified divacancy defective graphene.
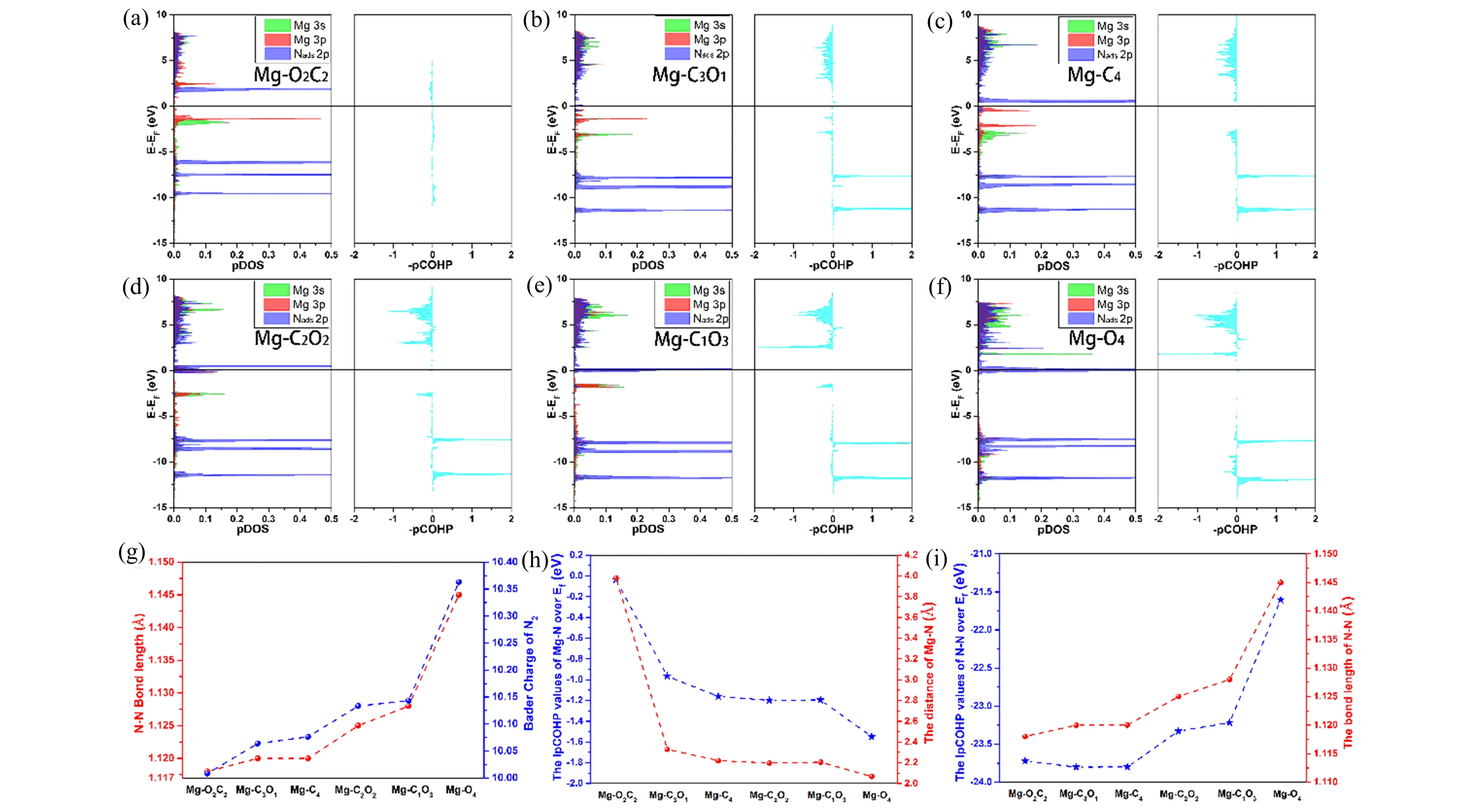
Figure 3. (a)-(f) Projected density of states (pDOS) and projected crystal orbital Hamilton populations (pCOHP) for the Nads atoms of *N2 directly bonded to the metal atoms of the six models. The relationship between the integrated pCOHP values up to the Fermi level and the bond length of (g) Mg-N, (h) N-N.(i) The relationship between the Bader charge and bond length of adsorbed N2 molecule.
However, we simultaneously noticed that the charge accumulation and depletion phenomenon of N2and catalyst in model Mg-O2C2is separated and their charges don’t overlap in space (Figure S3). Interestingly, the different site of coordinated O around Mg center in model Mg-O2C2and Mg-C2O2leads to distinct ability for N2adsorption. In fact, different coordinated structures determine the symmetry of the electronic structure of the Mg center and further exert influence on electronic orbit overlap between the Mg center and N2molecule. Figure 3 shows the projected density of states (pDOS) of the Nadsatoms in*N2directly bonded to the Mg atom. Compared Figures 3(a) to 3(d), the area of the 3s and 3p bands of the Mg atom in model Mg-C2O2overlapped with the 2p orbitals of Nadsis obvious larger than that in model Mg-O2C2, which indicates that the possibility of electrons exchange between Nadsand Mg enhances and stronger binding between Nadsand Mg-C2O2. Furthermore, the strength of bonding between*N2and Mg-C/O can be understood by projected crystal orbital Hamilton populations (pCOHP) analysis (Figure 3(a)-(f)). The positive and negative of integrated pCOHP values represent the bonding and anti-bonding respectively. As shown in the right part of Figure 3(a), the integrated area of the model Mg-O2C2is very small, which is corresponding to the result of pDOS analysis. The integrated pCOHP values up to the Fermi level for Mg-Nadsand Nads-N of six models are listed in Table S1, we thus could analyze the binding strength of N2adsorption in a quantitative way. The more negative of this value, the more anti-bonding state is above the Fermi level and more bonding state is under the Fermi level, which indicates stronger binding between adsorbent and catalyst. As the value for Mg-Nadsdecreases from model Mg-O2C2to Mg-O4, while the value for Nads-N increases, suggesting a stronger bonding between Nadsand Mg atoms and a weaker N-N bond. More importantly, as shown in Figure 3 (g) and 3(h), we further notice that the bond length of Mg-Nadsand Nads-N are also consequence with this trend. It is well-known that the change of N-N bond length determines the degree of nitrogen activation. Therefore, the model Mg-O4possesses the optimum ability for N2activation in above-mentioned six models. Simultaneously, we observe that the Bader charge of adsorbed N2molecule present the same tendency with the N-N bond length. This result provides full proof for that the activation mechanism of Mg-C/O structure coincides with the π back-donation process, which weakens the N-N bond and strengthen the metal-nitrogen bond at the same time.
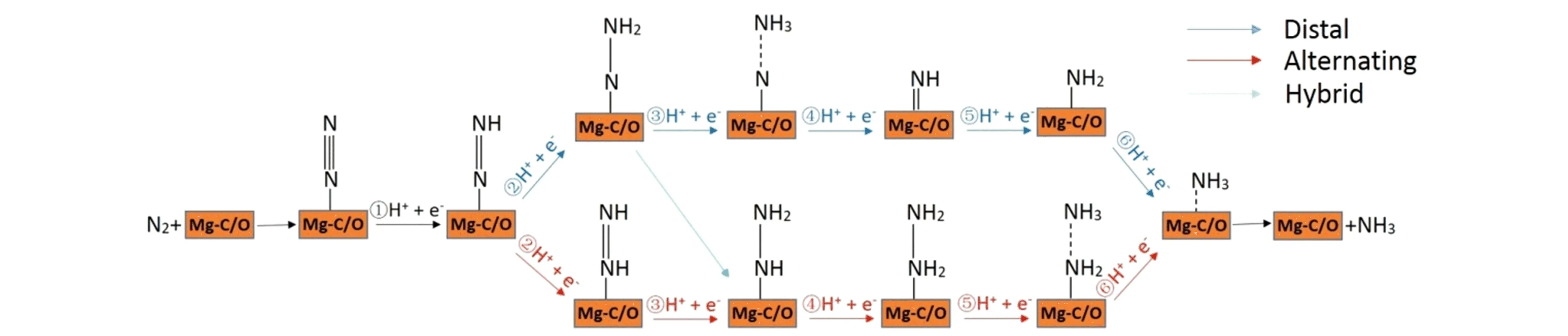
Figure 4. Schematic depiction of the three reaction mechanisms for the electrochemical NRR on Mg-C/O catalyst.
In addition, we further explore the HER activity of Mg-O4structure. The optimized structure of the adsorbed H on Mg-O4is shown in Figure S4. We can see the single H can also adsorbed by Mg atom. The Gibbs free energy diagrams indicate that H atom is more feasible adsorbed by Mg atom compared to N2molecule. However, due to the strong adsorption energy, the H is difficult to desorb from Mg atom to form H2. In the other words, once the N2molecule is adsorbed on the catalyst, the NRR is prior to occur. Thus, it is suggested that the selectivity of NRR would be higher than HER if less H atom in the electrocatalysis environment.
3.3 NRR catalytic mechanism on Mg-C/O
Typically, there are three possible reaction pathways for the N2end-on adsorption configuration on the Mg-O/C catalysts surface, i.e., distal, alternating, and hybrid pathways[39, 40]. As shown in Figure 4, in the mechanism of the distal pathways, the proton and electron pairs (H++ e-) are prone to interact with the terminal N atom at first, which results in the breakage of the N-N bond and the release of the first NH3molecule. After that, it sequentially attacks the other N atom to produce the second NH3. In comparison, the hydrogenation process of two N atoms would occur successively in the alternating pathways. In the hybrid pathway, the first two steps are the same as those in the distal pathway, and the following steps are along the alternating pathway. Given that the adsorption strength for N2of catalysts could directly affect the NRR catalytic performance, we firstly explored those three reaction pathways on the Mg-O4structure. In the Figure S5 and S6 are the top-view and side-view of optimized configurations along distal and alternating reaction pathways, respectively. We can observe that the structure of Mg-O4don’t collapse or distortion after the adsorption of various NxHyintermediates. Here, we did not list the hybrid pathway because the intermediate adsorbents of hybrid pathway is consistent with the other two ways.
As illustrated in Figure 5, the Gibbs free energy diagrams of above-mentioned three pathways on Mg-O4show both uphill and downhill steps, which means endothermic and exothermic steps coexist during the NRR process. Generally, the potential-determining step (PDS) of a reaction is the step with the maximum Gibbs free energy change (ΔGL) of endothermic step. Furthermore, the corresponding applied potential withΔGLto make all steps of reaction exothermic is named asUL. The Gibbs free energy change (ΔGL) is a very important indicator to evaluate the catalytic activity in thermodynamic perspective. In Figure 5(a), we can find that the first hydrogenation of the adsorbed N2(*N2+ H++ e-→*NNH) is endothermic with a free energy uphill of 1.04 eV. The similar phenomenon is also observed in the previous investigation of transition metal and main-group element based catalyst, such as Fe-N-C[36]and B-rich COFs[41]. In the following reaction procedures, the distal pathway undergoes another endothermic step with 1.35 eV Gibbs free energy (Figure 5(b)), which releases the first NH3molecule. In comparison, the Gibbs free energy of endothermic steps in alternating and hybrid pathways don’t overpass the first hydrogenation step. Thus, the potential-limiting step of distal mechanism is the third hydrogenation step with a NH3molecule release, while the first hydrogenation steps of alternating and hybrid pathways are their respective PDS. As the Gibbs free energy change (ΔGL) of the potential-determining step in distal mechanism is larger than that in alternating and hybrid pathways, the NRR prefers to follow the alternating and hybrid pathways on model Mg-O4structure.

Figure 5. (a) Gibbs free energy diagrams for the NRR on model Mg-O4 without applied potentials. Comparison of Gibbs free energy diagrams at 0 V (blue) and UL (red) via (b) distal, (c) alternating and (d) hybrid mechanisms.
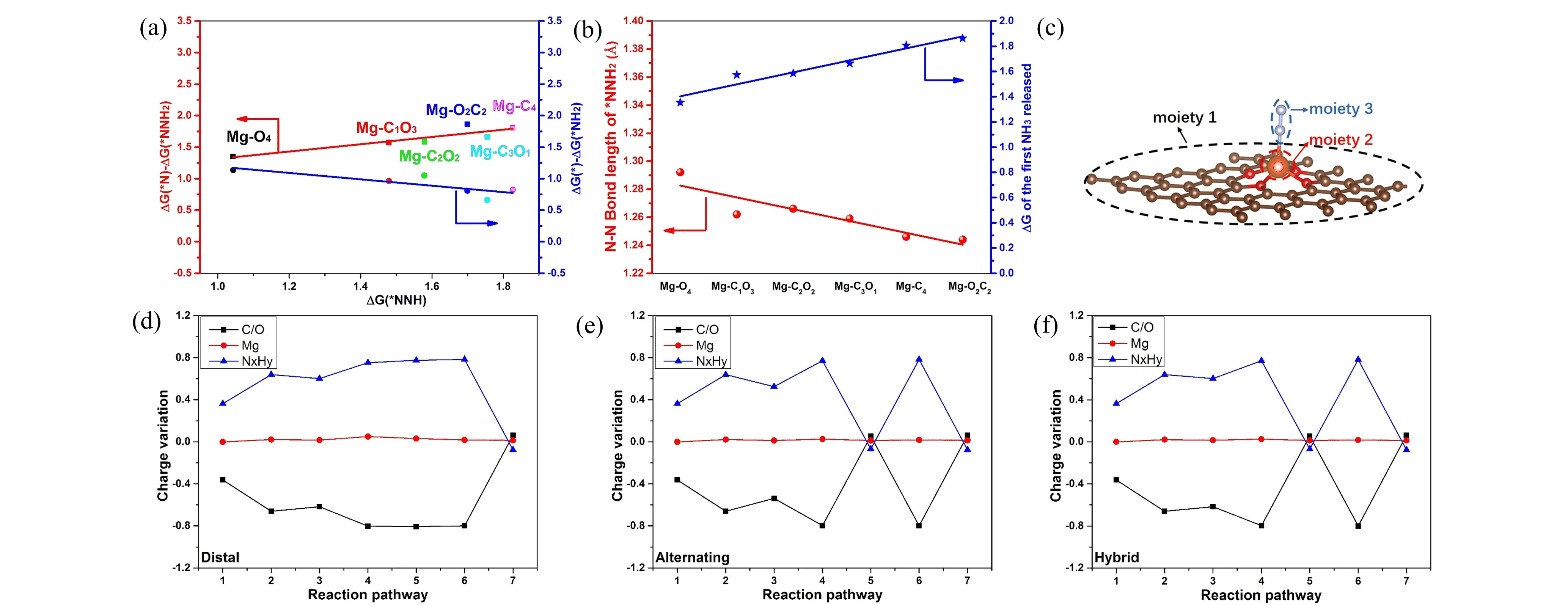
Figure 6. (a) The relationship of three uphill steps through distal pathway, left and right axis are the Gibbs free energy required for the first and second NH3 molecule release, respectively, x axis is the Gibbs free energy of the first hydrogenation of the adsorbed N2. (b) The trend between N-N bond and ΔG of second NH3 molecule release. (c) The three moieties of adsorbent structure. Charge variation of each moiety between the present step and the initial structure for Mg-O4 along the (d) distal, (e) alternating, and (f) hybrid pathway.
After that, we continued to explore those three NRR pathways on other five models. The adsorbed configurations of various NxHyintermediates on other models are illustrated in Figures S7-S16, meanwhile the corresponding Gibbs free energy diagrams are presented in Figure S16. We notice that all the distal pathways of the six models present three uphill steps: the first hydrogenation of N2and the release of two NH3molecules. Thus, the Gibbs free energy of those steps determines the reaction rate in distal mechanism. Interestingly, Figure 6(a) shows us two linear relationships: with the number of O atoms increasing, the Gibbs free energy required for the first and second NH3molecule release is linear scaling with the Gibbs free energy of the first hydrogenation of the adsorbed N2. However, their linear relationships are in opposite direction. This characteristic is different from the situation of transition metal surfaces, indicating that the Mg-O/C structures break the scaling relation between the energetics of uphill steps and could be candidate materials for ammonia synthesis[42]. Meanwhile, the first NH3release step requires larger Gibbs free energy (ΔG1) than the second NH3release (ΔG2), which means the former step determines the NH3yield rate. Furthermore, Figure 6(b) presents that the trend of N-N bond length of adsorbed intermediate NNH2in the distal pathway is inverse with the tendency ofΔG1. In other words, the longer of N-N bond length of*NNH2, the less energy needed for the first NH3release. In fact, a longer N-N bond length represents a weaker binding strength, thus energy required for the N-N cleavage decreases. As shown in Figures S18 (a)-(f), in the alternating pathway, the N-N length was elongated to 1.483 Å before the first NH3release step on all six Mg-C/O models, while the longest N-N bond length before the first NH3release step in the distal pathway is just 1.296 Å of model Mg-O4. Compared to distal pathway, alternating pathway is more favorable for NRR catalysis on the model Mg-C/O structures. Coincidentally, with the number of O atoms coordinated with Mg increasing, theΔGLof different models decreases in alternating pathway. Therefore, the model Mg-O4possesses the optimized NRR catalytic activity through the alternating mechanism.

Figure 7. Variations of temperature and energy against the time for AIMD simulations of Mg-O4, insert are top and side views of the snapshot of atomic configuration. The simulation is run for 10 ps with a time step of 2 fs at 300 K.
To gain further insights into the catalytic effect of the Mg-C/O on the N2-fixation, we performed the charge population analysis. As illustrated in Figure 6(c), each adsorption structure is divided into three moieties: O-modified graphene (moiety 1), Mg atom (moiety 2), and the adsorbed NxHyintermediates (moiety 3). Here, we defined the charge variation as the Bader charge difference between each moiety in the present step and that of the initial Mg-C/O structure without adsorbents. The charge variation at step 1 represents the mutual transferred charges between the three moieties at N2adsorption step. Figure 6 (d)-(f) are the charge variation diagrams of the distal, alternating, and hybrid pathways in Mg-O4, respectively. Impressively, the charge variation of Mg atom presents little fluctuation during the whole reaction process of all the three pathways. Meanwhile, the charge variation between O-modified graphene (moiety 1) and the bounded NxHyintermediates (moiety 3) is negative correlation. Superficially, the electrons for activating NxHyintermediates seem to originate from the O-modified graphene and have nothing to do with the Mg atom. In fact, the O-modified graphene substrate gains 1.67 electrons from the Mg atom after forming Mg-O4structure and the NxHyintermediates get 0.77 electrons at most during the whole NRR process. Therefore, it’s reasonable to believe that the valence electrons of Mg atom redistribute between the O-modified graphene and NxHyintermediates during the whole reaction procedure. In addition, as shown in Figure S19, the 4O-modified graphene model without the Mg atom anchoring presents no adsorption ability for N2molecule. Thus, the Mg atom is indispensable for NRR reaction in Mg-C/O structure. From above analysis, we can identify the role of each moiety in Mg-O/C structure: the 4O-modified graphene acts as an electron reservoir ascribing to the high charge density and the Mg atom is the active site for N2-fixation and the transmitter for the electron transfer, and the collaboration between them contributes to the system to be a novel catalyst for N2-fixation.
The thermal stability of the Mg-O4structure was evaluated by using AIMD simulations at 300 K. As shown in Figure 7, there is no significant geometrical distortion for Mg-O4during 10 ps simulation, indicating the relatively high stability. Due to the high stability of the catalyst, we envision that the Mg-O4structure could possess full advantages in terms of N2fixation at ambient conditions.
4 Conclusions
In summary, we have designed the main-group s-block metal single atom electrocatalyst, Mg-O4, with excellent activity for N2fixation. The mutual charge transfer between N2and Mg-O4confirms the “acceptance-donation” process, which is similar to the traditional metals for the capture and activation of N2. The subsequent N2reduction process prefers to occur through the alternating mechanism due to the longer bond between two N atoms before releasing the NH3molecule. Moreover, our computed result indicates the number of O coordination atom plays an important role for the catalytic activity of Mg atom. In addition, this catalyst exhibits a high thermal stability under ambient environment, which is suitable for sustainable NH3production.
Supplementarydata
Supplementary data are available at J. Univ. Sci. Tech. China online.
Acknowledgments
This study was supported by the National Natural Science Foundation (51772283, 22072140),the Fundamental Research Funds for the Central Universities (WK2060000032) and the Hong Kong Scholars Program (XJ2019022). The DFT calculations were completed on the supercomputing system in the Supercomputing Center of the University of Science and Technology of China.
Conflictofinterest
The authors declare no conflict of interest.
Authorinformation
YANG Kang and WANG Changlai are co-first authors.
YANGKangreceived his PhD in Materials Physics and Chemistry from University of Science and Technology of China in 2020. His research mainly focuses on preparation and applications of nanomaterials in the field of inorganic small molecule electrocatalysis.
WANGChanglaireceived his PhD in Materials Physics and Chemistry from University of Science and Technology of China in 2019. He is currently a postdoctoral fellow under the Hong Kong Scholars Scheme at City University of Hong Kong. His research focuses on electrochemical energy conversion and storage.
CHENQianwang(corresponding author)received his PhD from University of Science and Technology of China in 1995. He is currently Full Professor at the Hefei National Laboratory for Physical Sciences at the Microscale and the Department of Materials Science and Engineering, University of Science and Technology of China. His research mainly focuses on studies of several aspects of solid-state materials, including self-assembly synthesis, catalysis, and energy storage of nano-scale materials. He has published more than 300 papers in most distinguished international journals, such asNatureCommunications,JournaloftheAmericanChemicalSociety,AngewandteChemieInternationalEdition,AdvancedMaterials,Energy&EnvironmentalScience, andPhysicalReviewLetters.

- 中国科学技术大学学报的其它文章
- OER电催化剂的结构重构及同步辐射研究
- Sequence-dependent hybridizability of DNA-monoconjugated nanoparticles: Kinetic complexity unveiled by a dimerization assay
- A novel amidoxime modified polyethylene nanofibrous membrane with high uranium adsorption capacity
- Source apportionment of heavy metals in soil of Guangzhou: Comparison of three receptor models
- Summer thermal performance study on pipe-embedded PCM composite wall in existing buildings