
土著菌群与蚯蚓肠道菌群共享核心物种协同抵御灭蚁灵胁迫研究①
2022-01-24 13:41朱国繁应蓉蓉邓绍坡孔令雅钱家忠孙明明
土壤 2021年6期
朱国繁, 应蓉蓉,邓绍坡,孔令雅,钱家忠 *,孙明明,朱 瑜,叶 茂
土著菌群与蚯蚓肠道菌群共享核心物种协同抵御灭蚁灵胁迫研究①
朱国繁1, 应蓉蓉2,邓绍坡2,孔令雅2,钱家忠1 *,孙明明3,朱 瑜4,叶 茂5 *
(1合肥工业大学资源与环境工程学院,合肥 230009;2生态环境部南京环境科学研究所,南京 210042;3南京农业大学资源与环境科学学院,南京 210095;4海南大学政治与公共管理学院,海口 570208;5中国科学院南京土壤研究所,南京 210008)
采集实际场地中不同浓度梯度(0 ~ 27.7 mg/kg)灭蚁灵污染土壤,设置蚯蚓土培试验,采用高通量测序技术,分析蚯蚓肠道菌群和土著菌群的结构和功能;通过MetagenomeSeq分析、LEfSe(linear discriminant analysis effect size)分析和随机森林(random forests)分析以及网络分析,识别其核心类群。发现:①灭蚁灵胁迫下蚯蚓肠道菌群相较土著菌群的结构、组成受到的扰动更显著(<0.05);②不同浓度梯度灭蚁灵胁迫下,蚯蚓肠道菌群和土著菌群组合存在稳定的核心物种,主要为气单胞菌属()、黄杆菌属()、盖勒氏菌属()、微枝形杆菌属()、地杆菌属()、亨氏菌()、;③这些核心种群在互作网络的平均度中心性、紧密中心性、特征向量中心性分别为136.72、0.44、0.52,均高于非核心种群的对应指标(91.52、0.42、0.33),表明核心种群具有更高的网络连通性,且此类核心种群具有碳氮转化和农药降解能力,说明蚯蚓肠道菌群和土著菌群可以通过共享核心物种与其他微生物联系紧密,具备发挥农药降解代谢的潜在功能,实现协同抵御土壤中灭蚁灵胁迫。
灭蚁灵;土著菌群;蚯蚓肠道菌群;核心物种
灭蚁灵是典型的有机氯农药,在2004年被纳入国际公约《斯德哥尔摩公约》(Stockholm Convention)作为管控对象。我国在履行斯德哥尔摩国际公约以及“退二进三”“退城进园”等政策深入实施的过程中[1],遗留大量灭蚁灵污染场地,造成部分表层土壤中灭蚁灵浓度甚至可达61.9 mg/kg[2-3],存在较大的环境风险,严重威胁人类健康和生态安全。
针对此类农药污染场地,我国已陆续开展大量修复工作,其中微生物修复技术因其环境友好、高效等特点而被广泛运用[4]。土壤微生物是生态系统过程中的关键驱动因素,包括土壤养分循环、植物生长和生物地球化学过程[5]。蚯蚓作为土壤环境中生物量最大的土壤动物之一,参与土壤环境各种代谢过程,对维持土壤内稳态环境具有重要意义[6]。目前,针对土著菌群和蚯蚓肠道菌群在污染土壤环境体系中的研究,主要为土著菌群或蚯蚓肠道菌群对土壤污染物的降解作用,以及在该环境中单独发挥的生物功能,很少涉及土著菌群和蚯蚓肠道菌群组合的功能分析。微生物群落的功能分析较为常见的方法是构建互作网络来识别核心种群,剖析核心种群特征揭示整个微生物群落的生态功能[7]。目前一些包括差异分析在内的统计学方法常被用于分析土壤和肠道微生物群落样本组,因此,此类方法是识别核心种群的有效方法[8]。
本研究以土著菌群和蚯蚓肠道菌群为研究对象,使用高通量测序技术,测定不同浓度灭蚁灵胁迫下土壤和蚯蚓肠道中微生物群落,探究不同浓度灭蚁灵胁迫下,土著菌群和蚯蚓肠道菌群的演变过程;通过3种统计学分析方法:MetagenomeSeq分析、LefSe (linear discriminant analysis effect size) 分析和随机森林(random forests)分析,获知样本可操作分类单元(operational taxonomic units,OTU)的具体分类位置和显著程度,筛选具有分类学特征或规律的差异物种,作为本试验微生物群落的核心物种;通过网络分析,剖析核心种群在灭蚁灵胁迫土壤环境中的生态功能。本研究结果可为探明土著菌群与蚯蚓肠道菌群共享核心物种在抵御土壤中有机氯农药毒害胁迫的协同机制,提供新的科学认识。
1 材料与方法
1.1 供试生物和污染土壤
供试威廉腔环毛蚓()购自江阴市郊区附近农场,选择生长良好,表皮无损伤,有明显环带,体重在3 g ± 0.5 g的成年蚯蚓。
采集江苏省江阴市青阳镇某灭蚁灵污染场地表层土(0 ~ 20 cm),自然风干、混合均匀后,使用气相色谱仪测定获取农药浓度信息[9]。选取土壤样本中不同浓度梯度(T0,0 mg/kg;T1,1.2 mg/kg;T2,5.4 mg/kg;T3,27.7 mg/kg)灭蚁灵污染土壤作为试验土壤。
1.2 蚯蚓土培试验
采购的成年蚯蚓使用清水清洗后,清肠24 h,无菌水洗净。称量T0、T1、T2、T3土壤各500 g(每组3个平行),每份土壤投放10条清肠的威廉腔环毛蚓,置于透气黑色花盆(直径15 cm,高20 cm)中培养,称重,进行为期14 d的土培试验。试验期间每天添水至初始质量,直至试验周期结束。培养结束后,不同处理组中均无蚯蚓死亡,收集不同处理中的土壤和存活的蚯蚓,取肠道内容物充分混匀,–80 ℃冷藏。T0、T1、T2、T3处理组中蚯蚓肠道内容物和土壤样本分别记为E_T0、E_T1、E_T2、E_T3和S_T0、S_T1、S_T2、S_T3(T0为对照组)。根据Ye等[9]的检测方法,测定培育周期前后不同处理组土壤基本理化性质、农药浓度变化以及蚯蚓肠道对农药的富集系数,结果如表1所示。
1.3 高通量测序
①DNA的提取:用DNeasy PowerSoil试剂盒(QIAGEN)提取土壤和蚯蚓肠道内容物中微生物细胞的DNA,–80 ℃下冷藏;②引物设计:根据微生物核糖体RNA和特定基因片段序列中的保守区域,合成带有Barcode的特异引物对样本的16S rRNA V4区域进行扩增,前端引物为515F (5′-GTGCCAGCM GCCGCGGTAA-3′),反向引物为806R (5′-CCG TCAATTCMTTTRAGTTT-3′);③双端测序:采用Illumina公司的TruSeq Nano DNA LT Library Prep Kit制备测序文库,使用MiSeq测序仪进行双端测序;④DADA2序列去噪:调用qiime cutadapt trim-paired切除序列的引物片段,弃去未匹配引物的序列,通过qiime dada2 denoise-paired调用DADA2进行质控、去噪、拼接、去嵌合体,得到50 943 ~ 81 094条序列;⑤进行物种分类学注释:选用Greengenes数据库采用QIIME2的classify-sklearn算法[10],即对于每个OTU的代表序列,在QIIME2软件中使用默认参数,使用预先训练好的Naive Bayes分类器进物种注释;⑥OTU抽平:采用稀疏(Rarefaction)的方法,通过从每个样本中分别随机抽取最低样本序列量的95%,计算物种多样性和组成[11]。
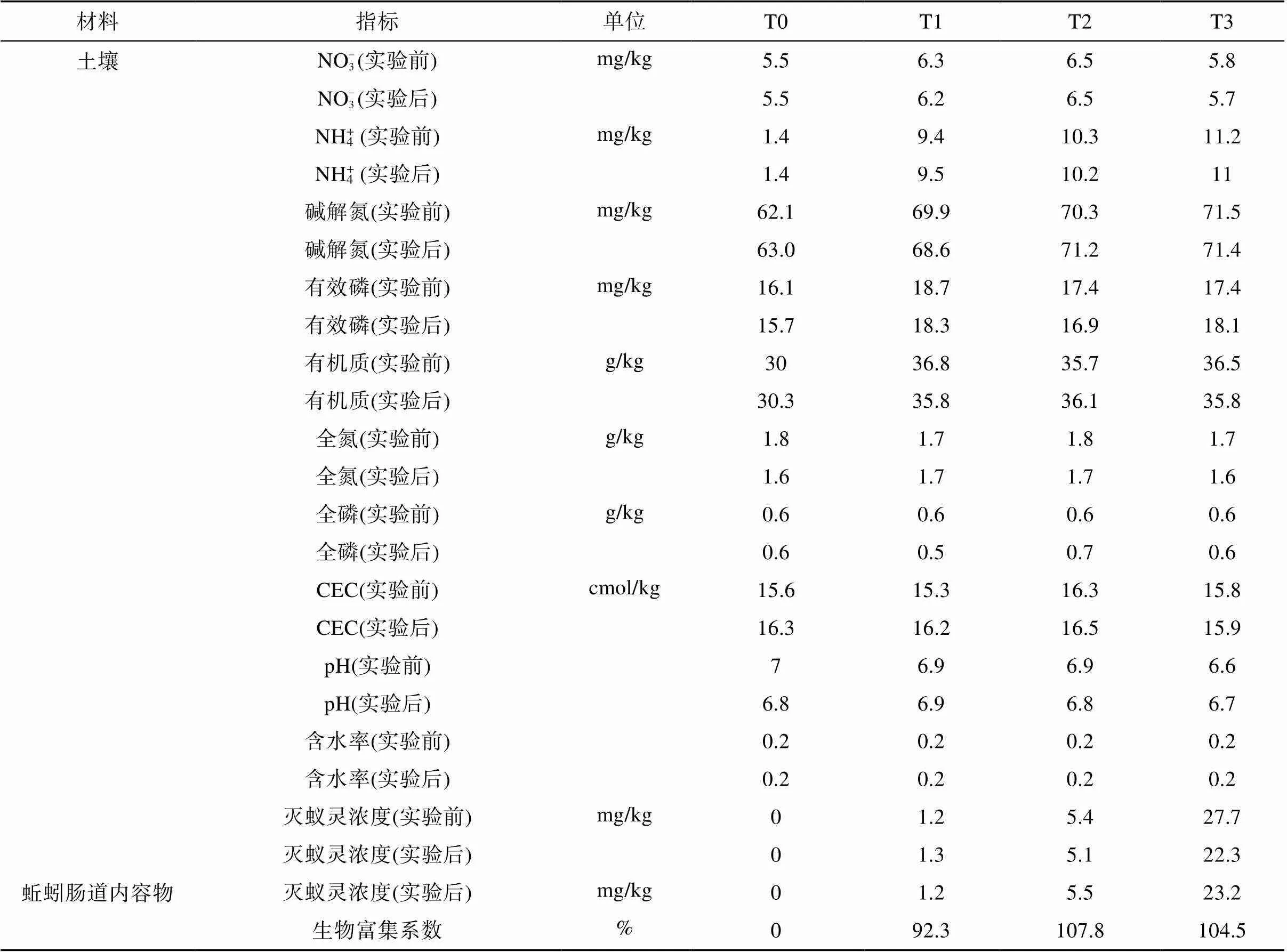
表1 土壤基本理化性质、农药浓度变化及蚯蚓肠道对农药的富集系数
1.4 功能潜能预测
①将已知微生物基因组的16S rRNA基因序列进行对齐,构建进化树,并推断它们共同祖先的基因功能谱;②将16S rRNA特征序列与参考序列对齐,从而构建新的进化树;③使用Castor隐藏状态预测算法,依据进化树中参考序列所对应的基因家族拷贝数,推测特征序列的最近序列物种,进而获得其基因家族拷贝数;④结合各样本的特征序列丰度,计算各样本的基因家族拷贝数;⑤将基因家族“映射”到MetaCyc和KEGG数据库中,使用MinPath推断代谢通路的存在,获得各样本中代谢通路的丰度数据。
1.5 MetagenomeSeq分析
使用获取的OTU数据,依据分组情况,每两组配对,调用fitFeatureModel函数使用zero-inflated log-normal model对每个OTU的分布进行拟合,并使用该模型的拟合结果判别土著菌群和蚯蚓肠道菌群中不同物种的差异显著性。该分析在R程序中进行(metagenomeSeq包)。
1.6 LEfSe分析
使用获取的OTU数据,将非参数的Kruskal- Wallis以及Wilcoxon秩和检验,与线性判别分析(linear discriminant analysis,LDA)效应量(effect size)相结合对所有物种分类水平丰度同时进行差异分析,寻找土著菌群和蚯蚓肠道菌群之间稳健的差异物种。该分析在R程序中进行(ggplot2包)。
1.7 随机森林分析
使用获取的OTU数据,以及分类水平在属的分类单元丰度表,调用“qiime sample-classifier classify- samples-ncv”命令进行巢式分层交叉检验分析(10倍交叉检验)。该分析在QIIME2 (2019.4)中进行。
1.8 网络分析
根据物种丰度特征(属水平)在R程序(psych包)中生成Pearson相关性矩阵,在软件Gephi-0.9.2中绘制T0-T1、T0-T2、T0-T3处理组蚯蚓肠道菌群和土著菌群共现网络图。
1.9 双因素方差分析
采用SPSS(IBM SPSS statistics 24)和R程序(Version 3.5.0)对数据进行统计分析,<0.05为有统计学显著差异意义。
2 结果
2.1 不同浓度灭蚁灵胁迫下土著菌群和蚯蚓肠道菌群的结构和组成
不同浓度梯度灭蚁灵处理组培育14 d前后,土壤中农药浓度、有机质、pH等理化性质未发生显著变化(表1)。通过高通量测序技术发现在土著菌群和蚯蚓肠道菌群中,均检测出古菌和细菌群落,但土著菌群和蚯蚓肠道菌群结构和组成差异显著(图1)。土著菌群以黄杆菌属(,22.3%)、气单胞菌属(,7.1%)、希瓦氏菌属(,2.3%) 为优势属;蚯蚓肠道菌群以贪铜菌属(,8.0%)、微枝形杆菌属(,3.9%)、伯克霍尔德菌属(,2.4%)为优势属(图1)。样本中土著菌群包含9 314个OTU,蚯蚓肠道菌群包含6 134个OTU,其中1 554个为共有OTU。土著菌群的OTU种类多于蚯蚓肠道菌群,而且其丰富度指数(Chao1、Observed_species)、多样性指数(Shannon、Simpson、Faith’s PD)和均匀度指数(Pielou’s evenness)也均高于蚯蚓肠道菌群(表2)。此外,不同浓度梯度灭蚁灵胁迫下,蚯蚓肠道菌群相比较土著菌群,其结构组成变化更显著(<0.05),尤其是T3处理组中微生物群落变动幅度更为明显。
2.2 土著菌群和蚯蚓肠道菌群的功能路径预测
土著菌群和蚯蚓肠道菌群在MetaCyc第一层次通路均以“生物合成(biosynthesis)”通路丰度最高,分别为60.9% 和61.1%;第二层次通路以“氨基酸生物合成(amino acid biosynthesis)”通路丰度最高,分别为14.3% 和15.6%(图2)。土著菌群和蚯蚓肠道菌群在KEGG第一层次通路均以“代谢(metabolism)”通路丰度最高,分别为81.7% 和81.5%;第二层次通路以“碳水化合物代谢(carbohydrate metabolism)”通路丰度最高,分别为13.0% 和13.5%(图3)。因此,不同浓度梯度灭蚁灵胁迫下,蚯蚓肠道菌群和土著菌群具有相似的功能通路丰度分布特性。
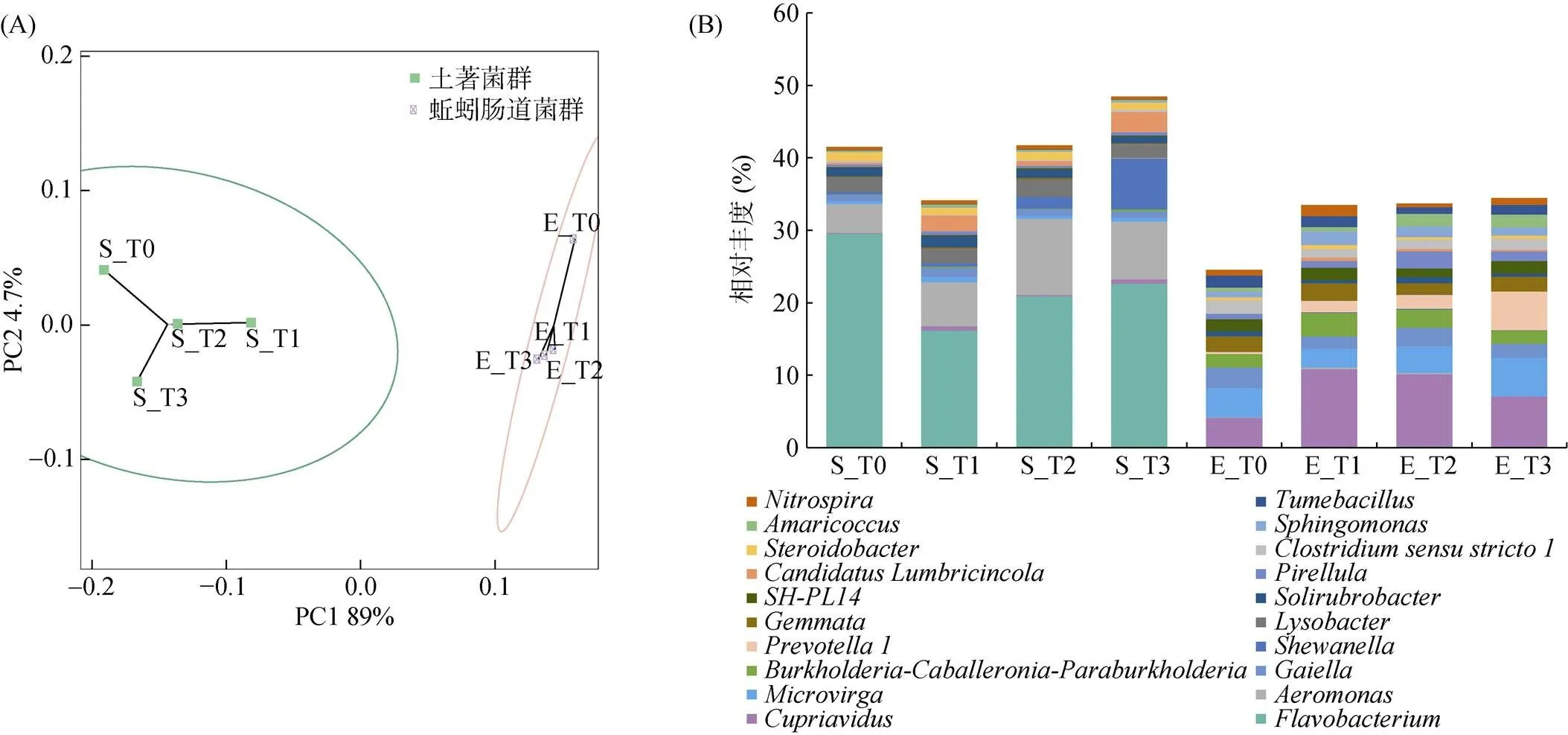
(图A为样本组的PCA分析。每个点代表一个样本(取平行组均值),不同颜色的点指示不同样本组。两点在坐标轴上的投影距离越近,表明这两个样本间的物种丰度组成在相应维度中越相似,椭圆形虚线圈为95% 置信椭圆(即该样本组100个样本中会有95个落在其中);图B为蚯蚓肠道菌群和土著菌群相对丰度前20位的属。横坐标为不同的样本组,纵坐标为对应的相对丰度值)

表2 土著菌群和蚯蚓肠道菌群的多样性指数
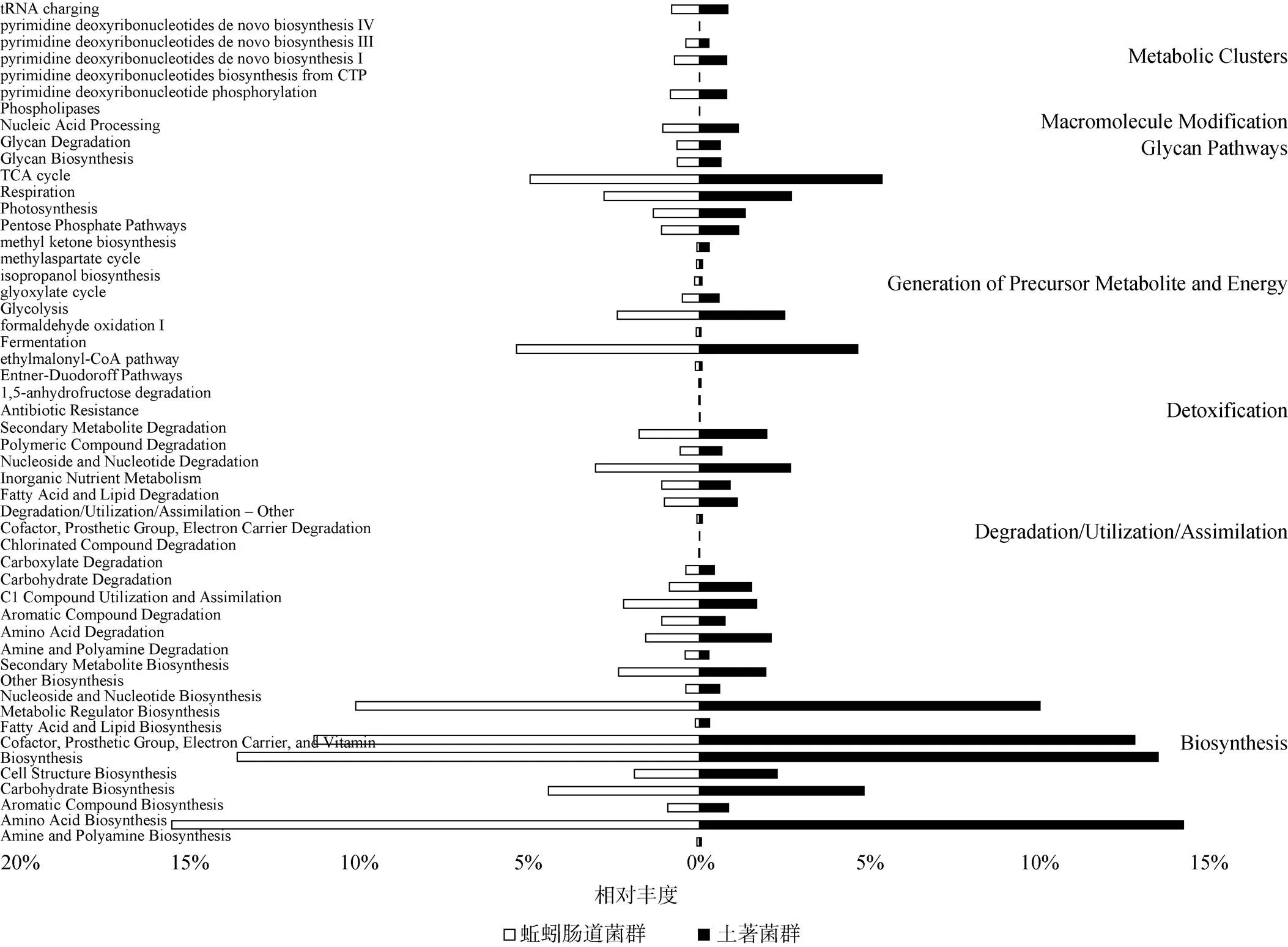
(横坐标为功能通路的丰度(平均相对丰度);纵坐标左侧为MetaCyc第二分类层次的功能通路,右侧为该通路所属的第一层次通路)
2.3 核心物种的识别
MetagenomeSeq分析基于不同样本组物种(相对丰度)在统计上的显著差异,筛选不同分类水平中具有富集趋势的物种;LEfSe分析强调寻找分组之间稳健的差异物种,不局限于对不同样本分组中的群落组成差异进行分析,还可以深入到不同的子分组中,挑取在不同子分组中表现一致的微生物类群;随机森林分析能够深入挖掘微生物之间复杂的非线性相互依赖关系,筛选对样本分析具重要性的微生物。本研究根据这3种分析方法,共筛选出51种核心物种(属),主要属于拟杆菌门(Bacteroidetes)、变形菌门(Proteobacteria)、放线菌门(Acfinobacteria)。其中7种为共同筛选物种,分别为:气单胞菌属()、黄杆菌属()、盖勒氏菌属()、微枝形杆菌属()、地杆菌属()、亨氏菌()、。土著菌群中核心物种以黄杆菌属(,22.3%)、气单胞菌属(,7.1%)、溶杆菌属(,1.5%)为优势属;蚯蚓肠道菌群中核心物种以贪铜菌属(,8.0%)、微枝形杆菌属(,3.9%)、普氏菌属(,12.3%)、盖勒氏菌属(,2.3%)为优势属(图4)。此类土著菌群和蚯蚓肠道菌群核心微生物同样存在于有机氯农药污染土壤环境中,虽然其相对丰度有所变化,但均具有发挥农药降解和有机质代谢潜能,是土壤微生物群落重要的成员[12-15]。不同浓度梯度灭蚁灵处理组中,土著菌群和蚯蚓肠道菌群的核心物种变化趋势略有差异(图5)。整体上,土著菌群核心种群随农药浓度的升高其相对丰度呈现先下降后上升趋势,蚯蚓肠道菌群核心种群随灭蚁灵浓度升高其相对丰度逐渐升高。

(横坐标为功能通路的丰度(平均相对丰度);纵坐标左侧为KEGG第二分类层次的功能通路,右侧为该通路所属的第一层次通路)
2.4 核心物种的网络特性及功能
统计T0-T1、T0-T2、T0-T3处理组蚯蚓肠道菌群和土著菌群共现网络图中核心物种的度中心性(degree centrality)、紧密中心性(closeness centrality)、特征向量中心性(eigenvector centrality),发现核心物种的平均度中心性、紧密中心性和特征向量中心性系数为136.72、0.44、0.52,均高于非核心种群的91.52、0.42、0.33(表3)。
2.5 土著菌群和蚯蚓肠道菌群的交互作用
对土著菌群和蚯蚓肠道菌群进行可重复双因素方差分析(表4),单独考虑蚯蚓肠道菌群的作用,其>0.05,对不同处理组中微生物群落的多样性高低影响较小;单独考虑土著菌群的作用,其<0.05,对不同处理组中微生物群落的多样性高低影响较大;土著菌群和蚯蚓肠道菌群之间的交互作用对不同处理组中微生物群落的多样性高低影响显著(<0.05,交互作用下的值远大于crit)。

(图A为基于MetagenomeSeq分析的差异OTU的曼哈顿图。其中,上图,土著菌群样本组上调;下图,蚯蚓肠道菌群样本组上调。以上两图分别采用OTU在分组A或B出现频次大于等于0.5作为条件,对MetagenomeSeq分析结果进行过滤。纵坐标为–log10(adj-P value)值,差异越显著,Y轴位置越高。坐标系内每个圆点或圈代表1个OTU,大小代表其相对丰度,以log2(CPM/n)为单位(CPM: 每百万拷贝数;n:样本数量)。虚线分隔了显著差异与不显著的OTU,显著差异的点用彩色圆点或圆环标志,不显著的用灰色圆环表示,该分组内显著上调的用彩色实心圆点显示。图中,圆点的颜色标识其门水平名称并标注于图底部(显著上调的点数前10位的门);对显著上调的点数排在前15位的目,添加灰度背景,且该目的名称标注于图顶部。图B为基于LEfSe分析的线性判别分析效应值核心物种柱状图。横坐标为组间具有显著差异的分类单元(属),纵坐标则以条形图直观地展示各分类单元的线性判别分析对数得分值。分类单元按照得分值大小进行排序,以此描述它们在样本分组中的特异性。长度越长表明该分类单元的差异越显著,条形图的颜色指示了该分类单元所对应的丰度最高的样本分组。图C为随机森林分析筛选核心物种热图。筛选随机森林分析中重要性大于0.01的物种(属))
3 讨论
3.1 土著菌群和蚯蚓肠道菌群之间存在物种和功能的传递
灭蚁灵处理前后,土壤的理化性质、农药浓度和蚯蚓肠道的农药富集系数变化并不显著,可能与灭蚁灵难降解特性、半衰期长以及培养周期较短有关[16-17]。但不同处理组中土著菌群和蚯蚓肠道菌群结构和组成有明显差异(图1),主要与土壤和蚯蚓肠道环境差异相关[18]。土著菌群和蚯蚓肠道菌群在T0组中共享物种种类(属水平)占总物种类别的23.6%,在灭蚁灵处理组中(T1、T2、T3)上升为73.1%。其中土著菌群增加60.2% 的新物种,主要为变形菌门(Proteobacteria)、放线菌门(Acfinobacteria)、绿弯菌门(Chloroflexi);蚯蚓肠道菌群增加61.5% 的新物种,主要为变形菌门、厚壁菌门(Firmicutes)、酸杆菌门(Acidobacteria)(图6)。这些微生物通过蚯蚓的进食活动进入蚯蚓肠道,又以蚓粪的形式进入土壤中,加强土著菌群和蚯蚓肠道菌群物种层面的沟通交流,携带有不同功能基因的微生物在土壤和蚯蚓肠道中传播,从而实现两种微生物群落之间功能的传递,尤其是较低浓度(T1、T2组)灭蚁灵胁迫土壤环境中这种功能传递作用更加活跃。土著菌群和蚯蚓肠道菌群在土培过程中(T1、T2和T3处理组),出芽菌属()、膨胀芽胞杆菌属()、气单胞菌属()、等相对丰度升高(图1),而这些微生物被证实与土壤碳氮代谢和农药降解有关[19-21]。土著菌群和蚯蚓肠道菌群均能够降解有机农药、石油烃、多氯联苯等污染物,主要通过共代谢作用或产生相关酶(如漆酶、细胞色素P450单加氧酶、脱氢酶、脱卤酶等)增强土壤中污染物的生物降解[15,22]。此外,具有抵御有机氯农药的功能微生物在蚯蚓肠道定殖参与污染物的降解,并通过蚓粪的形式进入土壤环境中,刺激土著菌群中有益微生物的繁殖[18, 20],驱动土著菌群和蚯蚓肠道菌群功能演变(图2、图3),进而加速消减土壤污染物。土著菌群和蚯蚓肠道菌群在代谢有机氯农药、改善土壤环境质量的过程中形成良性循环体系,提高了土壤生态系统服务职能,加快土壤生态功能多样性和稳定性的自恢复。
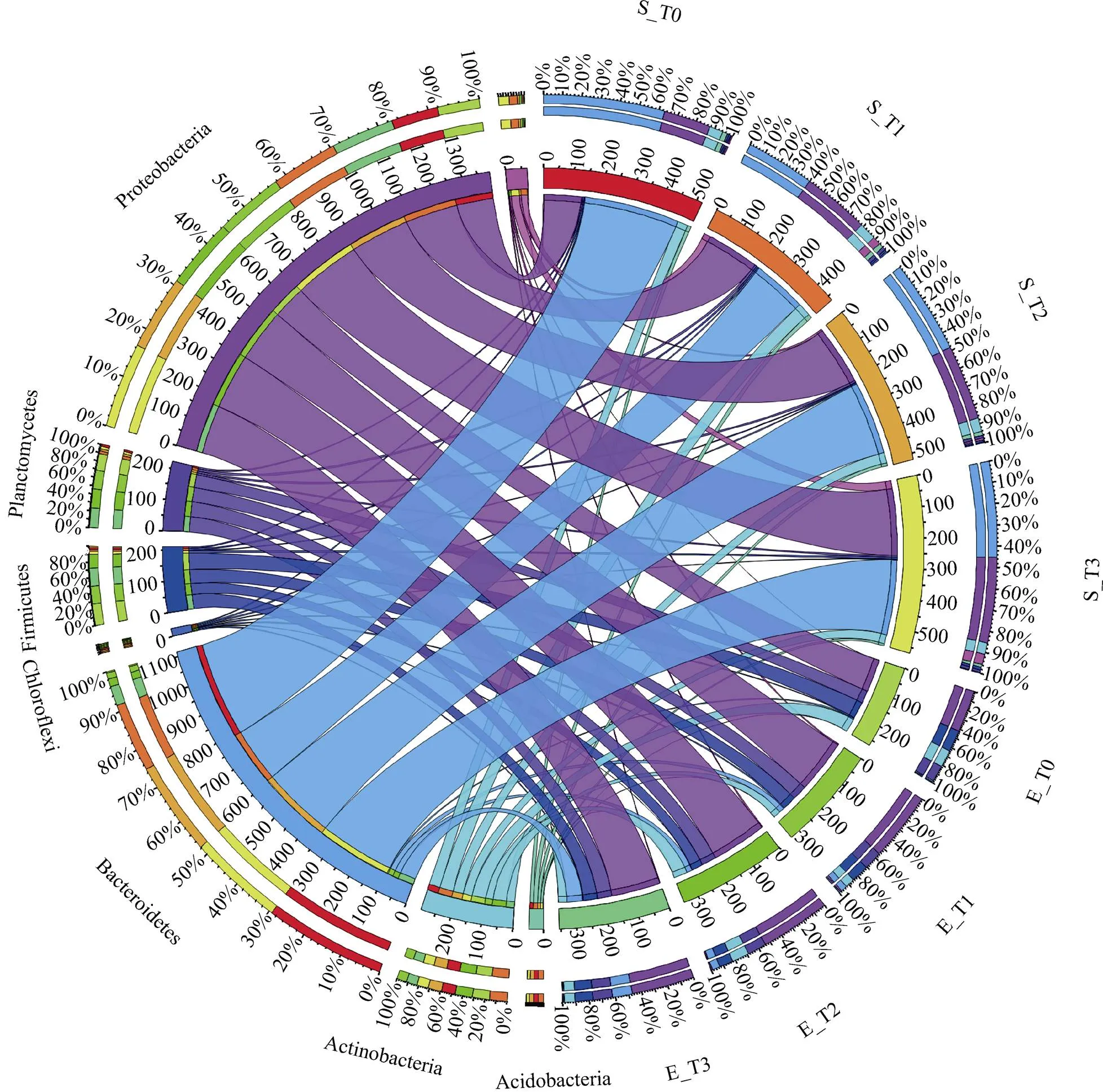
(筛选的核心微生物(门)在不同处理组中的相对丰度分布,内径为不同微生物的相对丰度(扩大1 000倍),外径为不同微生物在对应处理组中的比例;不同微生物的名称和处理组代码标记于最外圈)
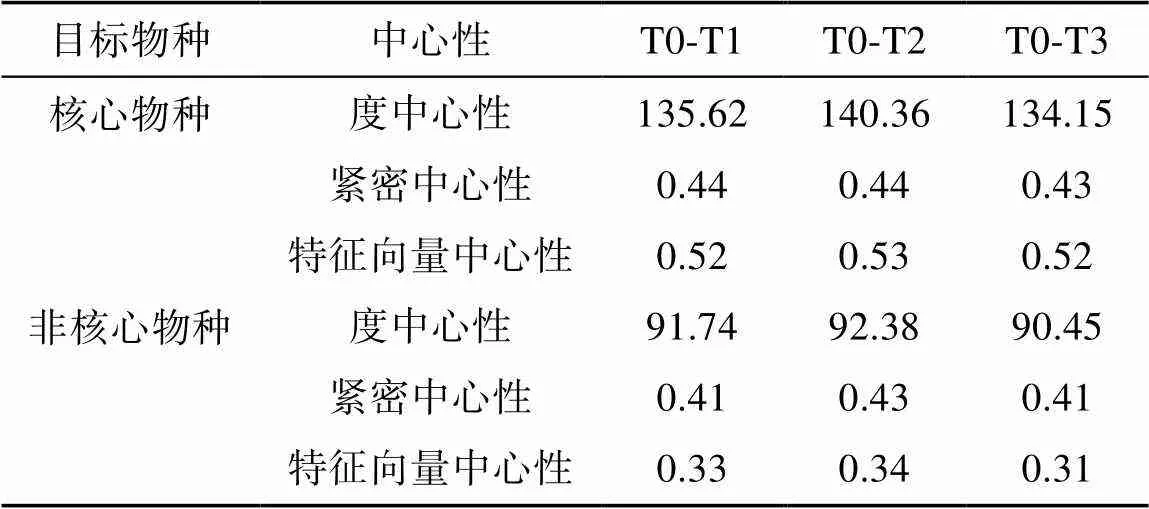
表3 土著菌群和蚯蚓肠道菌群互作网络的中心性
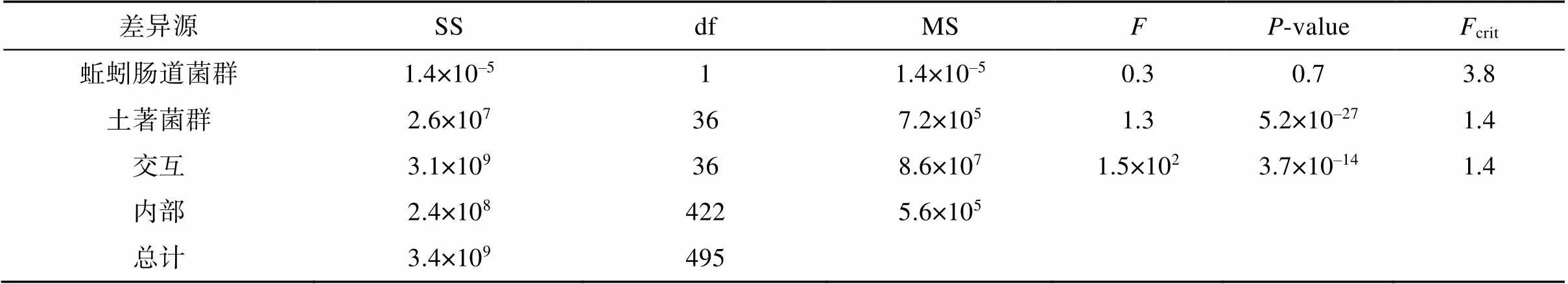
表4 可重复双因素方差分析
注: SS,离均差平方和;df,自由度;MS,均方;,统计量(处理均方/误差均方);crit,的临界值。
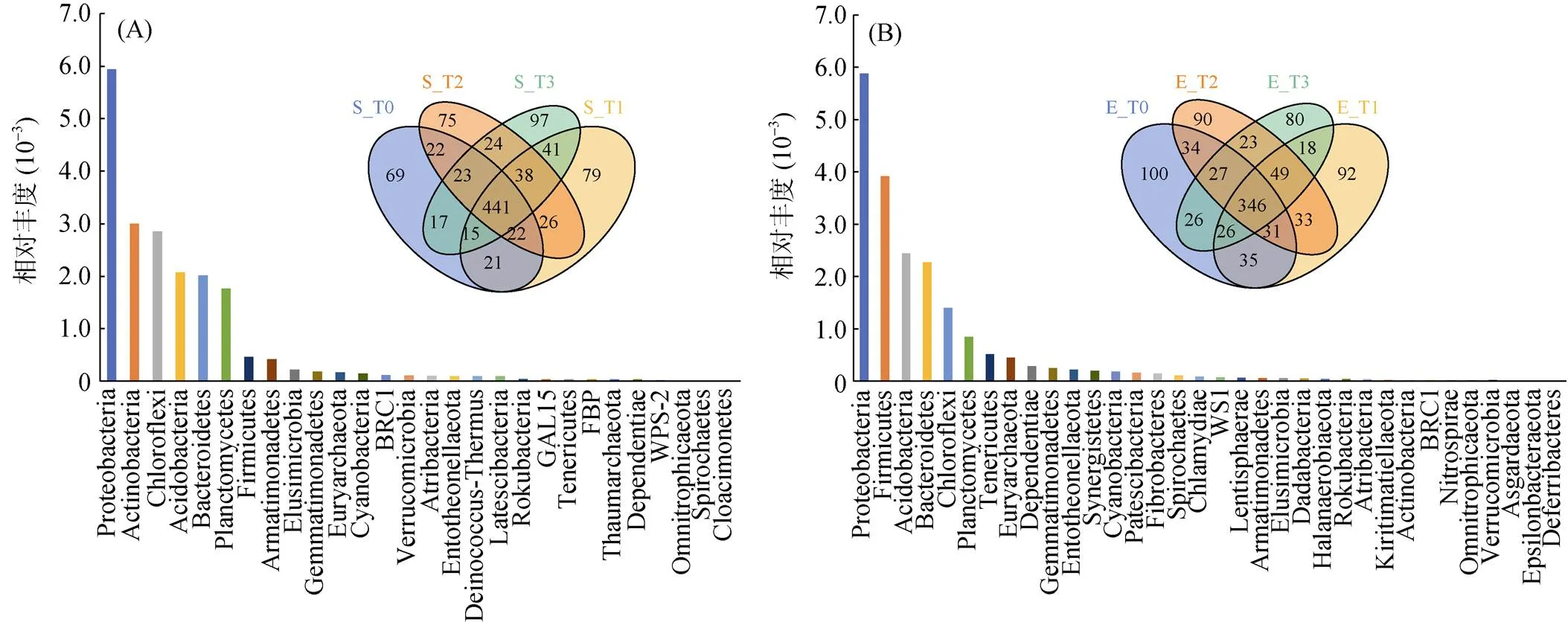
(图A:不同处理组土著菌群物种类别及新增物种分布;图B:不同处理组蚯蚓肠道菌群物种类别及新增物种分布。韦恩图表示土著菌群或蚯蚓肠道菌群在不同处理组中共有和特有物种的数目,S_T0、S_T1、S_T2、S_T3和E_T0、E_T1、E_T2、E_T3依次为T0、T1、T2、T3处理组中的土著菌群和蚯蚓肠道菌群样本组;柱状图表示T1、T2、T3实验组中新增物种的平均相对丰度,横坐标为物种名称(门分类水平),纵坐标为相对丰度值)
3.2 核心种群是土著菌群和蚯蚓肠道菌群应对农药胁迫的关键分类单元
土壤环境中不同微生物群落的演变趋势与土壤环境质量息息相关。研究人员常通过对土著菌群或蚯蚓肠道菌群的多样性、生物量、呼吸速率、特征微生物的丰度变化等指标进行分析,来评估土壤环境风险[23]。然而,土壤微生物群落及其驱动的过程很大程度取决于微生物之间的相互作用[24]。本研究中,不同浓度灭蚁灵胁迫(T0-T1、T0-T2、T0-T3)下,蚯蚓肠道菌群和土著菌群互作网络中存在稳定的高度连接节点(属水平物种),具有较高的网络拓扑特征(度中心性、紧密中心性、特征向量中心性),而且这些类群的结构和网络拓扑属性不随农药浓度升高而发生明显变化。较高的拓扑特征,表示微生物之间紧密的互作关系,丰富的降解路径和较强的生态位过滤特性(niche filtering)[25]。因此,蚯蚓肠道菌群和土著菌群互作网络中存在稳定的高度连接物种,可能具有较宽的生态位,还被证实与土壤中养分循环和农药代谢过程有关[19-21, 26]。如核心微生物黄杆菌属具有抵御有机氯农药,降解土壤中低浓度五氯苯酚(浓度为30和50 mg/L),参与有机质代谢的能力,还具有固氮功能[19];不动杆菌可使用杀虫剂作为唯一碳源进行共代谢[21];气单胞菌能够降解土壤中的有机氯农药[18]。此外,微生物互作网络具有无标度(scale-free)性质[27],表明蚯蚓肠道菌群和土著菌群中高度连接物种与其连接的大量物种共存形成小型微生物网络,通过影响高度连接物种将信息传递给微生物群落中其他成员,进而影响微生物群落的装配过程[24]。
本研究通过3种方法筛选出的核心物种(图4),在不同浓度灭蚁灵胁迫下微生物群落互作网络中具有较高的网络特性(表3),表明MetagenomeSeq分析、LEfSe分析和随机森林分析可作为识别微生物群落核心物种的方法。本研究中蚯蚓肠道菌群和土著菌群中核心物种在农药刺激作用下(T1、T2、T3),其相对丰度均有所上升,尤其是T3组,核心物种平均相对丰度相较之T0组显著升高,提高了9.8%(< 0.05)(图5)。不同微生物群落互作网络的核心物种通常具有能够适应该生境的生物功能,这种功能通常是在与群落中物种相互作用过程中发生[28]。因此,不同浓度灭蚁灵胁迫下,蚯蚓肠道菌群和土著菌群能够抵御灭蚁灵的毒害,正是由于这些稳定存在的核心物种,作为农药胁迫下蚯蚓肠道菌群和土著菌群之间传递信息的桥梁,联系微生物群落中大量其他物种,在微生物群落间运输碳氮代谢和有机氯农药代谢功能(可能是酶和代谢产物)[29]来抵御农药胁迫。根据双因素方差分析(表4),蚯蚓肠道菌群对不同处理组中微生物群落多样性影响较小,而土著菌群和蚯蚓肠道菌群之间的交互作用对不同处理组中微生物群落多样性影响显著,表明蚯蚓肠道菌群和土著菌群之间存在协同效应,可影响微生物群落结构和组成的演变,进而发挥抵御农药毒害功能,协助维护土壤环境微生物群落内稳态。
土著菌群和蚯蚓肠道菌群在土壤生物化学过程中存在物种的传递以及功能的转换,这种紧密的群落互作关系增强微生物群落结构的稳定性,提高微生物群落抵御逆环境的能力。其中核心种群,尤其是具有抵御农药胁迫(灭蚁灵)功能的核心种群,是连接土著菌群和蚯蚓肠道菌群的关键枢纽。因此,探明蚯蚓肠道菌群和土著菌群的菌群特性、协同演变趋势和互作模式对于维护土壤环境系统安全具有重要意义。
4 结论
本研究基于统计学方法和网络分析识别蚯蚓肠道菌群和土著菌群的核心种群,主要为气单胞菌属()、黄杆菌属()、盖勒氏菌属()、微枝形杆菌属()、地杆菌属()、亨氏菌()、,发现这些微生物不仅具有农药降解和碳氮代谢功能,也是维系蚯蚓肠道菌群和土著菌群紧密互作、协同抵御灭蚁灵胁迫的关键因素。本研究可以为深入阐释土著菌群和蚯蚓肠道菌群协同抵御农药污染胁迫过程和机制提供新的科学认知。
[1] 黄现民, 樊平, 闵建美, 等. 国际土壤农药污染研究进展与前沿——基于CiteSpace分析[J]. 土壤, 2021, 53(4): 764–770.
[2] Bonner M R, Alavanja M C R. Pesticides, human health, and food security[J]. Food and Energy Security, 2017, 6(3): 89–93.
[3] 赵玲, 滕应, 骆永明. 我国有机氯农药场地污染现状与修复技术研究进展[J]. 土壤, 2018, 50(3): 435–445.
[4] Navrátilová D, Tláskalová P, Kohout P, et al. Diversity of fungi and bacteria in species-rich grasslands increases with plant diversity in shoots but not in roots and soil[J]. FEMS Microbiology Ecology, 2019, 95(1): fiy208.
[5] Graham E B, Knelman J E, Schindlbacher A, et al. Microbes as engines of ecosystem function: When does community structure enhance predictions of ecosystem processes?[J]. Frontiers in Microbiology, 2016, 7: 214.
[6] Hu L, Xia M, Lin X H, et al. Earthworm gut bacteria increase silicon bioavailability and acquisition by maize[J]. Soil Biology and Biochemistry, 2018, 125: 215–221.
[7] Banerjee S, Schlaeppi K, van der Heijden M G A. Keystone taxa as drivers of microbiome structure and functioning[J]. Nature Reviews Microbiology, 2018, 16(9): 567–576.
[8] Zgadzaj R, Garrido-Oter R, Jensen D B, et al. Root nodule symbiosis indrives the establishment of distinctive rhizosphere, root, and nodule bacterial communities[J]. Proceedings of the National Academy of Sciences of the United States of America, 2016, 113(49): E7996–E8005.
[9] Ye M, Sun M M, Hu F, et al. Remediation of organochlorine pesticides (OCPs) contaminated site by successive methyl-β-cyclodextrin (MCD) and sunflower oil enhanced soil washing -L. cultivation[J]. Chemosphere, 2014, 105: 119–125.
[10] Bokulich N A, Kaehler B D, Rideout J R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2's q2-feature-classifier plugin[J]. Microbiome, 2018, 6(1): 90.
[11] Meziti A, Tsementzi D, Ar Kormas K, et al. Anthropogenic effects on bacterial diversity and function along a river-to-estuary gradient in Northwest Greece revealed by metagenomics[J]. Environmental Microbiology, 2016, 18(12): 4640–4652.
[12] Feng J Y, Jue S T, Zhu Y J, et al. Crop-dependent root-microbe-soil interactions induce contrasting natural attenuation of organochlorine lindane in soils[J]. Environmental Pollution, 2020, 257: 113580.
[13] Sun G D, Zhang X, Hu Q, et al. Biodegradation of dichlorodiphenyltrichloroethanes (DDTs) and hexachlorocyclohexanes (HCHs) with plant and nutrients and their effects on the microbial ecological kinetics[J]. Microbial Ecology, 2015, 69(2): 281–292.
[14] Kavitha V, Anandhan R, Alharbi N S, et al. Impact of pesticide monocrotophos on microbial populations and histology of intestine in the Indian earthworm(Kinberg)[J]. Microbial Pathogenesis, 2020, 139: 103893.
[15] Sun M M, Chao H Z, Zheng X X, et al. Ecological role of earthworm intestinal bacteria in terrestrial environments: A review[J]. Science of the Total Environment, 2020, 740: 140008.
[16] Jacquiod S, Puga-Freitas R, Spor A, et al. A core microbiota of the plant-earthworm interaction conserved across soils[J]. Soil Biology and Biochemistry, 2020, 144: 107754.
[17] Wang P, Zhang Q H, Zhang H D, et al. Sources and environmental behaviors of Dechlorane Plus and related compounds—A review[J]. Environment International, 2016, 88: 206–220.
[18] Lin Z, Zhen Z, Ren L, et al. Effects of two ecological earthworm species on atrazine degradation performance and bacterial community structure in red soil[J]. Chemosphere, 2018, 196: 467–475.
[19] Liu K H, Ding X W, Wang J J. Soil metabolome correlates with bacterial diversity and co-occurrence patterns in root-associated soils on the Tibetan Plateau[J]. Science of the Total Environment, 2020, 735: 139572.
[20] Govarthanan M, Ameen F, Kamala-Kannan S, et al. Rapid biodegradation of chlorpyrifos by plant growth-promoting psychrophilicsp. BT05: An eco-friendly approach to clean up pesticide-contaminated environment[J]. Chemosphere, 2020, 247: 125948.
[21] Lu X M, Lu P Z. Response of microbial communities to pesticide residues in soil restored with[J]. Applied Microbiology and Biotechnology, 2018, 102(1): 475–484.
[22] Hao Y Q, Zhao L X, Sun Y, et al. Enhancement effect of earthworm () on acetochlor biodegradation in soil and possible mechanisms[J]. Environmental Pollution, 2018, 242: 728–737.
[23] Ma L L, Xie Y W, Han Z H, et al. Responses of earthworms and microbial communities in their guts to Triclosan[J]. Chemosphere, 2017, 168: 1194–1202.
[24] Hoeffner K, Santonja M, Cluzeau D, et al. Epi-anecic rather than strict-anecic earthworms enhance soil enzymatic activities[J]. Soil Biology and Biochemistry, 2019, 132: 93–100.
[25] Lee K C, Archer S D, Boyle R H, et al. Niche filtering of bacteria in soil and rock habitats of the Colorado plateau desert, Utah, USA[J]. Frontiers in Microbiology, 2016, 7: 1489.
[26] Xu Y F, Teng Y, Wang X M, et al. Exploring bacterial community structure and function associated with polychlorinated biphenyl biodegradation in two hydrogen-amended soils[J]. Science of the Total Environment, 2020, 745: 140839.
[27] Ma B, Wang Y L, Ye S D, et al. Earth microbial co-occurrence network reveals interconnection pattern across microbiomes[J]. Microbiome, 2020, 8(1): 82.
[28] Niehaus L, Boland I, Liu M H, et al. Microbial coexistence through chemical-mediated interactions[J]. Nature Communications, 2019, 10: 2052.
[29] Zhou G W, Yang X R, Sun A Q, et al. Mobile incubator for iron(III) reduction in the gut of the soil-feeding earthwormguillelmi and interaction with denitrification[J]. Environmental Science & Technology, 2019, 53(8): 4215–4223.
Keystone Species of Soil Indigenous Flora and Earthworm Intestinal Flora Help to Resist Mirex Stress
ZHU Guofan1, YING Rongrong2, DENG Shaopo2, KONG Lingya2, QIAN Jiazhong1*, SUN Mingming3, ZHU Yu4, YE Mao5*
(1School of Resources and Environmental Engineering, Hefei University of Technology, Hefei 230009, China; 2 Nanjing Institute of Environmental Science, Ministry of Ecology and Environment, Nanjing 210042, China; 3College of Resources and Environmental Sciences, Nanjing Agricultural University, Nanjing 210095, China; 4School of Politics & Public Administration, Hainan University, Haikou 570208, China; 5 Institute of Soil Science, Chinese Academy of Sciences, Nanjing 210008, China)
In this study, soils with different mirex concentrations (0–27mg/kg) were collected to set up earthworm inoculation trials. High-throughput sequencing was used to analyze the structure and function of earthworm intestinal and soil indigenous microbial communities. Meanwhile, the keystone microbial taxa were identified through network analysis. The results obtained here found that 1) The structure and composition of the earthworm intestinal bacteria varied more significantly than those of the soil indigenous bacteria (<0.05); 2) The keystone bacteria remained stable in the worm gut and the soil despite varying mirex concentration, which was mainly consisted of,,,,,,; 3) The keystone bacteria had higher network connectivity than the rest bacteria, whose mean degree centrality, closeness centrality, eigenvector centrality values were 136.7, 0.44 and 0.52, respectively, clearly higher than those in the rest bacteria (91.52, 0.42 and 0.33, respectively). In addition, the keystone bacteria were closely involved in the carbon/nitrogen transformation and pesticide degradation, which not only protect the keystone bacteria from mirex toxicity, but might also assist other bacteria in relieving pesticide toxicity.
Mirex;Soil indigenous flora; Earthworm intestinal flora; Keystone taxa
X53
A
10.13758/j.cnki.tr.2021.06.020
朱国繁, 应蓉蓉, 邓绍坡, 等. 土著菌群与蚯蚓肠道菌群共享核心物种协同抵御灭蚁灵胁迫研究. 土壤, 2021, 53(6): 1250–1260.
国家重点研发计划项目(2018FYC1803100)、国家自然科学基金面上项目(42077106)和中国科学院青年创新促进会项目(2018350)资助。
通讯作者(qianjiazhong@hfut.edu.cn; yemao@issas.ac.cn)
朱国繁(1995—),女,安徽安庆人,硕士研究生,主要研究方向为农药污染土壤微生物修复。E-mail: zhuguofan@issas.ac.cn
猜你喜欢
当代水产(2022年8期)2022-09-20
昆明医科大学学报(2022年2期)2022-03-29
学校教育研究(2020年7期)2020-04-09
环球时报(2017-10-10)2017-10-10
作文通讯·初中版(2017年1期)2017-04-13
科技知识动漫(2017年1期)2017-02-06
创新作文(1-2年级)(2015年11期)2015-11-28
食品工业科技(2014年13期)2014-03-11
小哥白尼·野生动物画报(2009年11期)2009-12-21
