
基于ddRADseq 的马铃薯品种遗传多样性分析
2022-01-24 08:55:18单建伟索海翠刘计涛李成晨白建明李小波
广东农业科学 2021年12期
单建伟,索海翠,王 丽,安 康,刘计涛,李成晨,白建明,李小波
(1.广东省农业科学院作物研究所/广东省农作物遗传改良重点实验室,广东 广州 510640;2.云南省农业科学院经济作物研究所,云南 昆明 650000)
【研究意义】马铃薯为茄科(Solanaceae)茄属(Solanum)马铃薯组(Petota)草本植物,原产于南美洲安第斯高原[1]。马铃薯的植物分类学是一个不断更新和完善的过程,Spooner 等综合前人有关马铃薯表型、分子标记、系统发生、生殖生物学、倍性、核型等方面的研究将马铃薯组分类为107 个野生种和4 个栽培种[2]。对于栽培作物而言,通常在其起源地具有最丰富的遗传资源和遗传多样性,而在其传播过程中遗传多样性逐渐降低[3]。20 世纪20 年代,前苏联、英国、德国和美国学者先后赴南美洲考察和收集马铃薯野生种及栽培种种质资源,为马铃薯的育种和遗传学研究开创了新纪元[4]。当今世界范围内广泛种植的现代马铃薯品种(modern cultivar)是以马铃薯普通栽培种为基础,通过与其他地方栽培种或野生种杂交利用了有利基因的产物,使其能够适应更宽泛的生态条件和地理区域,但目前广泛种植的马铃薯品种遗传多样性依然很低[5]。广泛收集国内外马铃薯品种(系)资源,对其群体结构和遗传多样性进行分析评价,将为我国马铃薯遗传育种提供理论指导。
【前人研究进展】我国马铃薯的栽培历史并不长[6],对其育种起步也较晚,始于20 世纪30年代中末期,早期的马铃薯育种工作以引种为主,先后从英国、美国和前苏联等引进一批育成品种和杂交组合。由于马铃薯适应性广、产量高,新中国成立以后为了尽快恢复农业生产,国家有计划地组织调运种薯,扩大马铃薯种植面积,全国开展了马铃薯育种协作,真正开始马铃薯育种工作,促进了马铃薯在我国的迅猛发展,并相继育成了一批有代表性的马铃薯品种,如东农303、克新系列、陇薯系列等[7-8]。当前,我国马铃薯种植面积和总产量均超过全球的1/4,居世界首位。由于我国马铃薯育种工作起步较晚,所用亲本多引自欧洲和北美等国,对种质资源特别是野生种和地方栽培种的收集、研究、创新不够,因此我国栽培的马铃薯品种遗传多样性更低,许多品种之间同质性高,部分品种存在同种异名现象,仅靠表型差异难以分辨。
【本研究切入点】为了指导马铃薯遗传育种,有必要收集国内外马铃薯品种(系)资源,并对其遗传多样性进行评价。【拟解决的关键问题】本研究对收集的国内外马铃薯品种(系)资源,利用限制性酶切位点关联DNA 测序(Restrictionsite Associated DNA sequencing,RADseq)中 的ddRADseq 技术对其进行简化基因组测序,结合群体结构分析和遗传多样性分析对其进行群体结构和遗传多样性评价,开发马铃薯SNP-Panel,为马铃薯育种、品种鉴定提供理论依据。
1 材料与方法
1.1 试验材料
供试材料为广东省农业科学院作物研究所马铃薯研究室收集的185 份马铃薯品种(系),包含育成品种以及一些高代品系(表1)。这些材料分别来自我国的华南、华北、西南、华中地区以及国外的秘鲁和意大利。
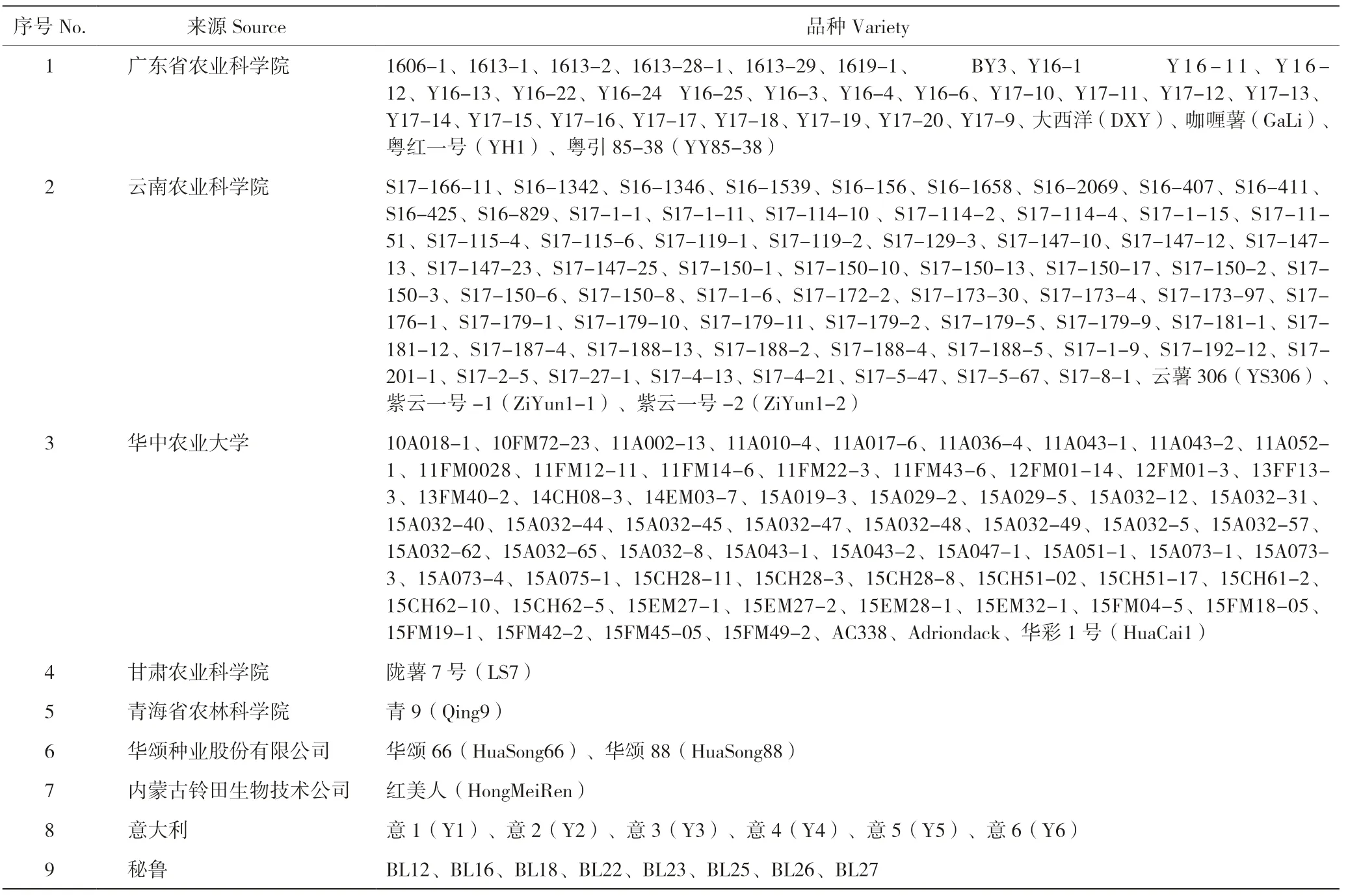
表1 马铃薯样品来源Table1 Sources of potato samples
1.2 试验方法
1.2.1 基因组DNA 制备 取每个马铃薯品种(系)的嫩叶,利用液氮速冻后储存于-80℃超低温冰箱,分离样品基因组DNA 后,利用琼脂糖凝胶电泳检测DNA 样品完整性及有无RNA 污染,并利用分光光度计对DNA 样品的质量和浓度进行定量检测。
1.2.2 文库构建、质控及测序 取约200 ng 基因组DNA 用限制性内切酶MseⅠ和SacⅠ(纽英伦生物技术有限公司)按照产品说明书进行完全酶切;酶切产物连接特异性接头,然后对连接产物进行纯化;用高保真聚合酶KOD-Plus-Neo(东洋纺生物科技有限公司)按照产品说明书进行PCR 扩增富集;将所有扩增产物进行低压电泳,切取300~400 bp 大小的电泳片段,用琼脂糖凝胶回收试剂盒(QIAGEN,德国)进行纯化;用分光光度计对纯化产物进行初步定量,用安捷伦2100 生物分析仪对文库插入片段大小进行检测;采用实时荧光定量PCR 手段对文库的有效浓度进行准确定量,有效浓度>2 nmol/L 为合格;最后用Illumina HiSeq 测序平台进行双末端测序。
1.2.3 测序数据过滤 测序得到的原始数据被称为raw read,含有测序接头序列或低质量的碱基。使用Cutadapt(Version 1.13)软件去除raw read中的接头序列,用Trimmomatic(Version 0.36)去除测序数据中的低质量碱基,得到clean read 用于后续分析;测序read 的长度必须大于50 bp。
1.2.4 参考基因组比对 使用BWA(Version 0.7.15-r1140)软件中的MEM 算法将测序数据与参考基因组(http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml)进行比对,得到SAM 格式的比对结果,然后使用 Samtools(Version 1.3.1)将其转换为BAM 格式,使用Picard(Version 1.91)软件中的SortSam 对BAM 文件中的序列进行排序,将处理后的BAM 文件用于覆盖度和覆盖深度统计以及变异位点识别。
1.2.5 变异位点检测及注释 利用GATK(Version 3.7)软件包中的HaplotypeCaller 模块对所有样品生成gvcf 格式文件,然后用GenotypeGVCFs 模块对所有样品进行SNP 和 InDel 变异检测。并按照测序深度≥5、所有样品基因型缺失率≤50%、稀有等位基因频率≥10%、杂合率≤50%、相对杂合率≤67%等标准对变异位点进行筛选,得到的变异位点为有效变异位点。用ANNOVAR(Version 2016Feb1)对变异位点进行注释。依据变异位点在参考基因组上的位置信息以及参考基因组中的基因位置信息,可得到变异位点在基因组中的区域(基因间区、基因区或CDS 区等)信息。
1.2.6 马铃薯群体结构分析 通过Structure 软件对群体结构进行分析,结果以ΔK值变化图和祖先堆叠图展示,可直观反映个体间的分类关系以及每个样品的“混血”程度。采用PLINK(Version v1.90p)软件对群体结构进行主成分分析(Principal Component Analysis,PCA)。PCA 散点图中,两个样品距离越远,表明遗传背景差异越大,而遗传背景相似的个体在PCA 散点图中聚类在一起。采用MEGA7(Version 7.0)软件中的邻接法(Neighbor-joining methods)构建系统发生树,并通过ggtree(Version 1.7.10)进行可视化处理。基于分子标记信息,利用Cervus 3.0 软件计算多态性信息含量(Polymorphism Information Content,PIC)、观测杂合度(Observed Heterozygosit,Ho)、期望杂合度(Expected Heterozygosity,He)等。
1.2.7 SNP 分子标记开发 在综合区分度和检测成本的基础上,结合SNP 标记在染色体上的位置信息以及多态性,选择120 个在染色体上分布相对均匀的SNP 标记,分别在SNP 位点上下游开发PCR 引物,PCR 产物长度介于150~350 bp 之间。
2 结果与分析
2.1 变异位点检测及注释
通过测序最终获得clean read 7.50×108条、2.04×1011个碱基,平均每个样品4.05×106条clean read,1.10×109个碱基。为了检测变异位点,将测序得到的clean read 与马铃薯基因组(http://solanaceae.plantbiology.msu.edu/pgsc_download.shtml)进行比对,并进行群体结构和遗传多样性进行分析。本研究在综合考虑可行性和经济性的基础上对测序覆盖度进行了控制,样品平均测序覆盖度为2.62%。利用GATK 软件包共检测到39 038 个有效变异位点,其中SNP 位点36 267个,InDel 位点2 771 个。SNP 位点在马铃薯12条染色体上的平均分布密度为50.02 个/Mb SNP位点,但在不同染色体上的的分布密度不均,其中10 号染色体上的SNP 位点密度最低(26.22 个/Mb),5 号染色体上SNP 位点密度最高(83.50个/Mb);InDel 变异位点在12 条染色体上的分布密度介于1.97~6.05 个/Mb 之间,同样10 号染色体分布密度最低,5 号染色体分布密度最高(表2)。同一条染色体不同区域的变异位点密度也存在较大差异,如1 号染色体10~20 Mb 范围内的变异位点密度最高,每100 kb 存在约90 个变异位点,而在70~90 Mb 范围内变异位点密度较低(图1)。
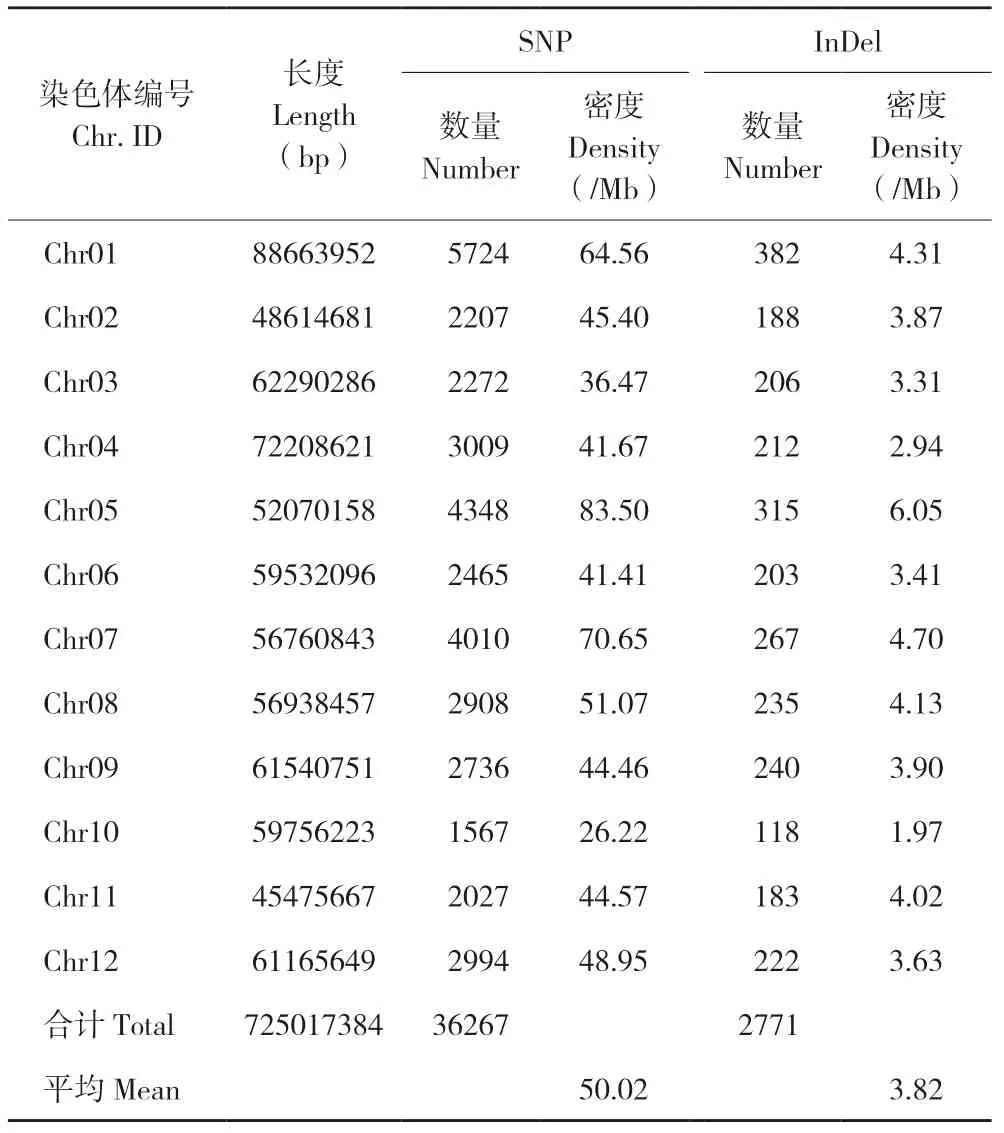
表2 变异位点检测统计结果Table 2 Detection and statistic results of variation sites
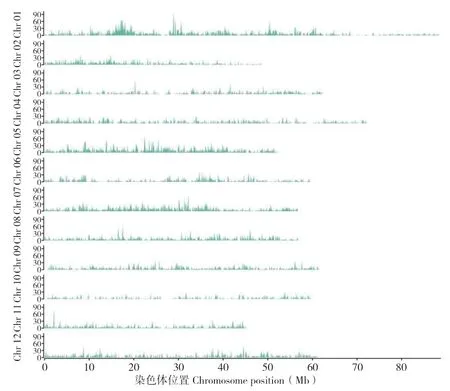
图1 变异位点在染色体上的分布Fig.1 Distribution of variation sites on chromosomes
利用ANNOVAR 软件结合变异位点在参考基因组和自身基因组上的位置信息对变异位点进行注释,结果(表3)显示,共有26 697 个SNP 位点位于基因间区,占所有SNP 位点的73.65%;1 392 个SNP 位点位于基因转录起始位点上游1 kb 范围内,1 314 个SNP 位点位于转录终止位点下游1 kb 范围内,116 个SNP 位点位于一个基因转录起始位点上游1 kb 范围内并同时在另一个基因的转录终止位点下游1 kb 范围内;6 729个SNP 位点位于基因内部,占所有SNP 位点的18.56%,其中3 172 个SNP 位点位于内含子区域,2 605 个SNP 位点位于外显子区;352 个SNP 位点位 于5′ UTR 区,597 个位 点位于3′ UTR 区;3 个SNP 位点位于可变转录本上。此外,共有1 761 个(63.57%)InDel 位点位于基因间区;308个InDel 位点位于基因编码区上下游1 kb 范围内;25.31%的InDel 位点位于基因内部,其中406 个位于内含子内,134 个位于外显子内;3′ UTR 和5′UTR 区域内的InDel 位点数分别为101、56 个;4个InDel 位点位于可变转录本内。
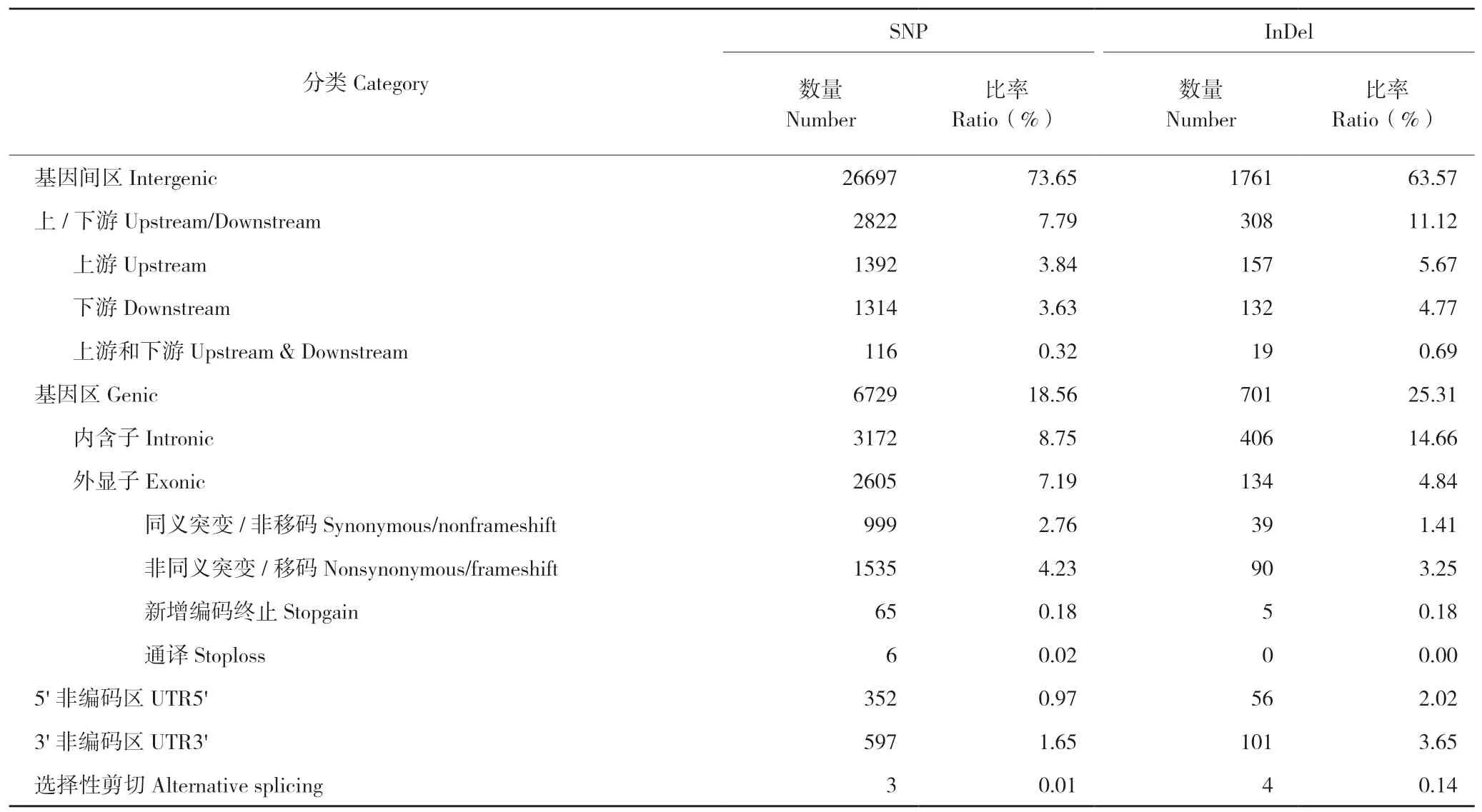
表3 注释的变异位点分布Table 3 Distribution of annotated variation sites
2.3 马铃薯群体结构分析
为了明确本研究所用马铃薯的群体结构,通过PCA 法对筛选得到的有效SNP 标记进行降维处理,从中提取关键信息将分子标记差异信息反映在二维坐标图上,坐标轴为对样品差异性解释度最高的特征向量,两个样品遗传背景差异越小,则样品分子标记位点信息越相似,反映在PCA 图中的距离越近。PCA 散点图显示,第一特征向量(PC1)和第二特征向量(PC2)可以将本研究所用群体区分为G1 和G2 两个亚群(图2A)。利用Structure 软件对群体结构进行分析,群体结构分析的ΔK值变化趋势图显示,当K=2 时对应的ΔK值最大,因而本研究群体的最佳亚群数为2,这一结果与PCA 分析结果一致,即将本研究群体分为两个亚群较为合适(图2B)。G2 亚群包含29 个品种(系)全部来自云南省农业科学院,分别为S17-192-12、S17-114-4、S17-188-13、S17-119-2、S17-188-4、S17-114-2、S17-166-11、S17-147-12、S17-114-10、S17-1-1、S17-176-1、S17-173-97、S17-4-13、S17-201-1、S17-173-4、S17-147-23、S17-187-4、S17-1-9、S17-4-21、S17-173-30、S17-1-11、S17-129-3、S17-27-1、S17-8-1、S17-1-15、S17-181-12、S17-172-2、S17-181-1、紫云一号-1;剩余的156 个品种(系)构成G1 亚群,包括来自我国华南、华北、华中、西南、西北等地的品种(系),以及引自秘鲁、意大利等国的品种(系)。祖先堆叠图(图2C)显示,两个亚群间存在明显基因交流,部分样本存在混血。群体系统进化分析结果(图2D)表明,所有G2 亚群中的29 个品种(系)聚类在一起且其分枝离圆心的距离最长,表明G2 亚群中的个体亲缘关系较近,同时G2 亚群中的个体在进化上积累了更多变异。
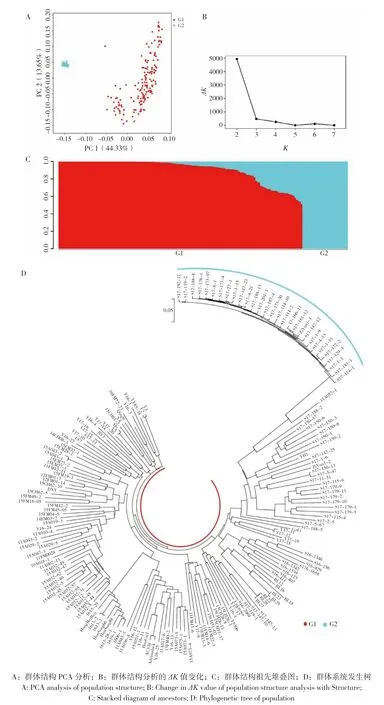
图2 马铃薯群体结构分析Fig.2 Analysis of potato population structure
PIC>0.5 为高度多态性[9],而本研究群体的平均PIC=0.3107,表明群体遗传多样性中等偏低。同时,群体的期望杂合度He=0.3932,观测杂合度Ho=0.1852,群体自交系数Fis=0.553,也表明群体遗传多样性偏低。
2.4 SNP 分子标记开发
靶向测序(target-seq)技术通过设计多重PCR 引物对目标区域DNA 片段进行特异性扩增,再通过双端标签区分不同样本,混样建库,进行多样本多位点高通量测序分析,具有信息量大、针对性强、低成本、效率极高、精准度高等特点,被广泛应用于分子标记辅助选择育种(Marker-Assisted Selection,MAS)、数量性状位点(Quantitative Trait Locus,QTL)精细定位、品种指纹分析、品种鉴定、基因编辑位点检测等领域。为了便于后续开展马铃薯亲缘关系鉴定、品种鉴别、群体遗传分析以及分子标记辅助选择育种,我们开发了包含120 个马铃薯SNP 标记的SNP-Panel,这些SNP 标记均匀分布在马铃薯的12 条染色体上(图3),并针对这120 个SNP 标记设计了分离目标DNA 区域的特异引物用于后续多样本高通量SNP标记检测。为了验证SNP 标记的有效性,选取了60 个SNP 标记,利用与所选SNP 标记对应的引物对46 份马铃薯品种(系)进行了靶向高通量测序,结果表明,60 个SNP 标记能够准确将46 份马铃薯品种(系)区分开。理论上,所选标记越多,越能区分更多的品种(系),因此需要在区分度和检测成本之间寻求平衡。因此在综合区分度和检测成本的基础上,本研究确定了SNP 标记的个数为120。
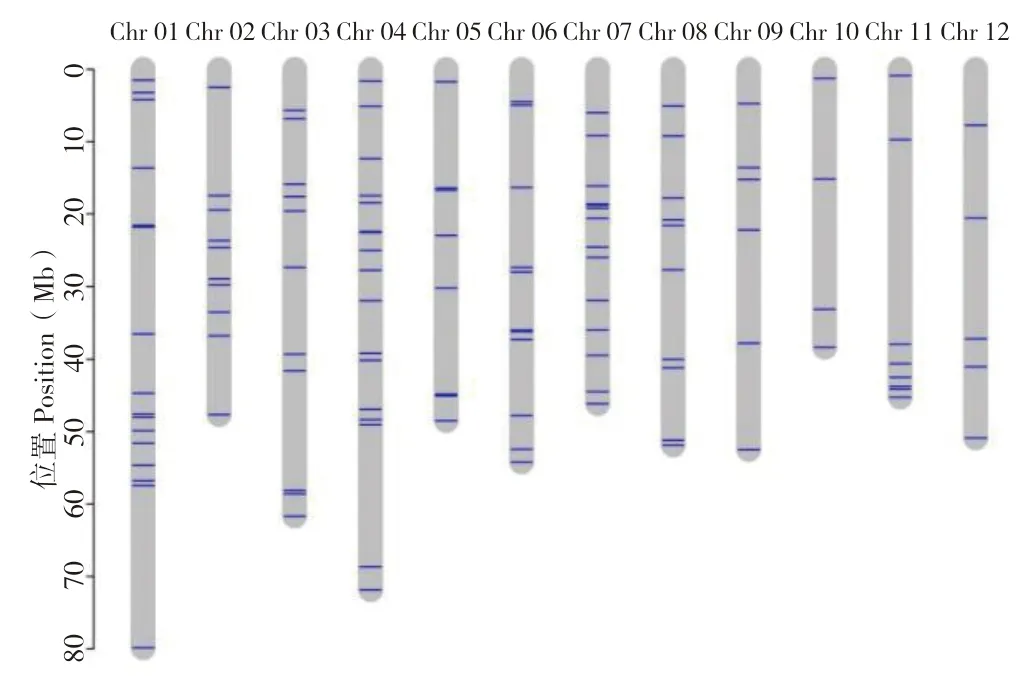
图3 SNP-Panel 在马铃薯染色体上的分布Fig.3 Distribution of SNP-Panel on potato chromosomes
3 讨论
RADseq 是在第二代测序技术基础上发展而来的一项基于全基因组酶切位点的简化基因组测序技术,该技术具有检测速度快、高通量、成本低等优点,可以检测出上千万的SNP 位点,能够满足群体结构分析、遗传图谱构建和QTL 定位等研究需求[10-11]。依据所使用的限制性内切酶的数量和种类,RADseq 技术可细分为Original RADseq、GBS、2b-RADseq、ezRAD、ddRADseq等技术方法。ddRADseq 技术由于检测准确、成本低、SNP 位点丰富等优点得到广泛应用[12-13]。本研究采用ddRADseq 技术对185 个马铃薯品种(系)进行了简化基因组测序分析。基于变异位点在各样品中的测序深度均不小于5、在所有样品中的缺失率不超过50%、低频等位基因频率不低于10%、杂合率不超过50%、相对杂合率不超过67%等标准共获得39 038 个高质量变异位点,其中SNP 位点36 267 个,InDel 位点2 771 个。高质量变异位点为后续马铃薯群体结构分析、SNP 标记Panel 开发、分子标记辅助选择育种、QTL 位点定位奠定了基础。
物种的遗传多样性越高对生存环境的适应性越强,抵御生物胁迫、非生物胁迫的能力越强,易于拓展其生存分布范围。本研究通过对来自不同地方的185 份马铃薯品种(系)的遗传多样性分析发现,多态性信息含量PIC=0.3107,处于中等偏低水平;群体期望杂合度He=0.3932,而观测杂合度Ho=0.1852,即Ho<He。这一结果表明这些马铃薯品种(系)的遗传多样性较低,即不同品种(系)间的同质性较高,这与前人研究结果较为一致[14-18]。为了提升马铃薯品种的遗传多样性,在未来的马铃薯育种工作中应加强对马铃薯种质资源的收集和创新,特别是对马铃薯野生种的利用。马铃薯种质资源丰富,包含107个野生种和4 个栽培种。原始栽培种和野生种含有众多调控马铃薯晚疫病、疮痂病、青枯病、病毒病、黑胫病抗性的等位基因,也含有能够调控马铃薯高淀粉和蛋白质含量、低还原糖含量、耐霜冻、休眠期、早熟等品质和农艺性状的优良等位基因[8]。加速对马铃薯种质资源的收集、评价、研究、创新和利用,有利于突破当前马铃薯育种的瓶颈[19-22]。
本研究结果表明,供试马铃薯种(系)间的遗传多样性较低,工作中也发现一些品种间表型差异很小,如果不借助分子标记技术,仅从表型差异很难区分。鉴于此,本研究还开发了包含120 个马铃薯SNP-Panel,这些SNP 标记均匀覆盖马铃薯12 条染色体,并针对这些SNP 标记设计了分离目标DNA 区域的特异引物,以用于后续进行多样本高通量靶向SNP 标记检测,为后续开展马铃薯亲缘关系鉴定、品种鉴别、群体遗传分析以及分子标记辅助选择育种奠定基础。
4 结论
本研究对收集的185 份马铃薯品种(系)种质资源进行简化基因组测序,鉴定了39 038个变异位点,其中SNP 位点36 267 个,InDel位点2 771 个;PCA 分析和Structure 分析均将供试的185 份马铃薯品种(系)划分为两个亚类;遗传多样性分析表明,群体多态性信息含量PIC=0.3107,期望杂合度He=0.3932,观测杂合度Ho=0.1852,自交系数Fis=0.553,群体遗传多样性较低;开发了覆盖马铃薯12 条染色体且包含120 个SNP 标记的SNP-Panel。本研究为后续进行马铃薯遗传多样性研究、分子标记辅助选择育种、分子身份证开发、品种鉴定、遗传图谱构建奠定了一定基础。
猜你喜欢
今日农业(2021年11期)2021-08-13 08:53:24
种子(2021年3期)2021-04-12 01:42:22
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
百科知识(2015年18期)2015-09-10 07:22:44
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
世界科学(2013年6期)2013-03-11 18:09:33
