
Effect of Shengmai Yin on the DNA methylation status of nasopharyngeal carcinoma cell and its radioresistant strains
2022-01-19 06:40ShiyLiuZhiyunWngDoqiZhuJiinYngDndnLouRuijioGoZetiWngAiwuLiYingLvQinFn
Shiy Liu,Zhiyun Wng,Doqi Zhu,Jiin Yng,Dndn Lou,Ruijio Go,Zeti Wng,Aiwu Li,Ying Lv,**,Qin Fn,*
aSchool of Traditional Chinese Medicine,Southern Medical University,Guangzhou,510515,China
bNanFang Hospital,Guangzhou,510515,China
Keywords:
Nasopharyngeal carcinoma
DNA methylation
Radiosensitization
Shengmai Yin
Peer review under responsibility of Xi’an Jiaotong University.
A B S T R A C T
Shengmai Yin(SMY)is a Chinese herbal decoction that effectively alleviates the side effects of radiotherapy in various cancers and helps achieve radiotherapy's clinical efficacy.In this study,we explored the interaction mechanism among SMY,DNA methylation,and nasopharyngeal carcinoma(NPC).We identified differences in DNA methylation levels in NPC CNE-2 cells and its radioresistant cells(CNE-2R)using the methylated DNA immunoprecipitation array and found that CNE-2R cells showed genomewide changes in methylation status towards a state of hypomethylation.SMY may restore its original DNA methylation status,and thus,enhance radiosensitivity.Furthermore,we confirmed that the differential gene Tenascin-C(TNC)was overexpressed in CNE-2R cells and that SMY downregulated TNC expression.This downregulation of TNC inhibited NPC cell radiation resistance,migration,and invasion.Furthermore,we found that TNC was hypomethylated in CNE-2R cells and partially restored to a hypermethylated state after SMY intervention.DNA methyltransferases 3a may be the key protein in DNA methylation of TNC.
1.Introduction
Nasopharyngeal carcinoma(NPC)is a malignancy affected by a variety of pathogenic factors as well as multiple genes.Radiotherapy has become the main curative treatment for NPC due to its radiosensitivity and unique anatomical location[1].In recent years,with lifestyle changes and improvements in medical technology,the incidence and mortality of NPC have significantly declined[2].However,some patients are resistant to irradiation,developing local recurrence or distant metastasis.Moreover,the large heterogeneity between individuals,even at the same TNM stage or under the same treatment,indicates that genetic differences may result in radiosensitivity variations[3].
Accumulating evidence indicates that radioresistant cancer cells that exhibit stem cell characteristics are the main cause of radiation resistance,with the mechanism reportedly being epigenetically regulated[4].DNA methylation is one of the currently known epigenetic mechanisms associated with radioresistance[5].Many studies have shown that radiation is related to changes in global DNA methylation,and that these changes may differ between tissues and even among strains of the same species.The mechanisms of changes in DNA methylation caused by radiation have not yet been determined;however,the effects of radiation on DNA methyltransferases as well as the changes in the methylation status of related genes in irradiated cells may be the most plausible mechanisms[6].Widespread aberrant methylation is often observed in NPC[7];nevertheless,whether changes in DNA methylation in NPC affect radiosensitivity remains unknown.A study indicated that 5-azacytidine enhanced radiosensitivity both in vitro and in vivo in NPC cells,which suggests that DNA methylation is indeed associated with radiosensitivity in NPC[8].
Tenascin-C(TNC)is a glycoprotein of the extracellular matrix(ECM)that participates in a wide range of cellular processes,including migration,invasiveness,and angiogenesis.High TNC expression has been observed in various cancers and is associated with poor prognosis in malignant tumors[9,10].Many factors influence the expression of TNC.A previous study found that in H9c2 cells,exposure to angiotensin II can reduce DNA methylation of the TNC promoter,and hence promote its expression[11].In addition,different studies have reported TNC induced by radiation;thus,although the mechanism is unknown,it may be related to tumor angiogenesis and metastasis after radiotherapy[12].
Radiosensitizers can effectively increase cancer radiosensitivity,but their safety and practicability in clinical trials remains to be demonstrated[13].Traditional Chinese Medicine(TCM)treatments can effectively reduce the side effects of radiotherapy[14];moreover,an increasing number of studies show that it can improve radiosensitivity[15,16].Shengmai Yin(SMY)is a Chinese herbal decoction traditionally used to treat the lack of Qi and Yin.Clinically,SMY can effectively alleviate the side effects of radiotherapy in various cancers,improve the quality of life of patients,and help achieve the desired effect of radiotherapy.Studies have shown that ginsenoside Rg3,an active ingredient of SMY,can sensitize colorectal tumors[17],non-small cell lung cancer[18],and esophageal carcinoma[19]to radiotherapy.These studies and practices have shown that SMY can be used as a potential radiosensitizer.
Although SMY and its active compound have a radiosensitizing effect of NPC as we have indicated in this paper,the mechanism is still need to be ulteriorly clarified.As mentioned above,DNA methylation may play a role in the radioresistance in NPC.Thus,we aimed to evaluate whether SMY can affect the DNA methylation profile of radioresistant strains in NPC as well as to assess the expression and methylation of TNC and its effect on radiosensitivity in NPC.
2.Materials and methods
2.1.Cell culture and reagents
The human NPC cell line CNE-2 was provided by Sun Yat-sen University(Guangzhou,China).The human NPC radioresistant cell line CNE-2R was established from CNE-2 by repeated exposure to radiation[20].The cells were cultured in 10% fetal bovine serum(Thermo Fisher Scientific,Waltham,MA,USA)diluted with RPMI-1640 medium(Thermo Fisher Scientific,Waltham,MA,USA)at 37°C in a humid incubator with 5% CO2.The cell concentration of SMY(Shengmai Injection,SuZhong Pharma,Taizhou,Jiangsu,China)was calculated as the total concentration(μg/mL)of active ingredients(Table S1).The cells were grouped as follows:CNE-2,CNE-2R,and CNE-2R+SMY(50μg/mL,48h).
2.2.DNA extraction
Three independent replicates were set for each treatment group,with pooled independent replicate samples from each group.The genomic DNA was extracted using the TIANamp Genomic DNA Kit(Tiangen,Beijing,China),according to the manufacturer's instructions.
2.3.Methylated DNA immunoprecipitation(MeDIP)array and data analysis
The MeDIP array and data analysis were performed by Aksomics Bio-tech(Shanghai,China).DNA was randomly divided into pieces ranging from 400 to 500 bp.The methylated DNA was immunoprecipitated with a 5-methylcytidine monoclonal antibody.The input DNA was labeled with Cy3-,whereas the immunoprecipitated DNA was labeled with Cy5-,and the two were then hybridized with Arraystar Human Block Array.Subsequently,the standardized data were analyzed using NimbleScan v2.5(Roche-NimbleGen,Madison,WI,USA)to identify methylation peaks.The difference in promoter methylation status among groups was compared through differential methylation enrichment peaks(DEPs).The criteria used by the NimbleScan algorithm in seeking DEPs were as follows:1)more than one group has a median(log2 MeDIP/input)≥0.3,and M’>0;2)more than half of the probes in a peak have a coefficient of variability not higher than 0.8 in both groups.The KEGG pathway and gene ontology(GO)analyses of differential genes were also performed.
2.4.Genomic methylation-specific PCR and bisulfite sequencing
Genomic DNA was extracted using the TIANamp Genomic DNA Kit(TIANGEN,Beijing,China).Bisulfite-treatment of DNA was performed according to the technique first developed by Herman et al.[21].Primers targeting TNC CpG islands were designed using Methprimer (https://www.urogene.org/methprimer),and all primer sequences are listed in Table S2.The genes were amplified using a touchdown multiplex PCR strategy,described as follows:initial denaturation at 94°C for 5min,and then proceeding to the first 19 cycles(94°C for 30 s,62°C for 30 s,and 72°C for 30s),with the annealing temperature decreased by0.5°Cpercycle until 53°C;then,performing the second 19 cycles(94°C for 30 s,53°C for 30 s,and 72°C for 1 min),with a final elongation at 72°C for 10 min,and the cycle terminated at 4°C.The PCR product was cloned into the pMD™18-T vector(Takara,Shiga,Japan)and eight positive clones were randomly chosen for bisulfite sequencing.
2.5.Small interfering RNA(siRNA)assay
SiRNA was provided by Ribobio(Guangzhou,China).The siRNA sequences for TNC and the sequences for negative control siRNA are shown in Table S3.SiRNA was transfected into CNE-2R cells using Lipofectamine 3000(Invitrogen,Waltham,MA,USA).Cells were collected for subsequent experiments 48 h after transfection.
2.6.RNA analysis by real-time PCR(RT-PCR)
Total RNA was extracted using TRIzol reagent(Thermo Fisher Scientific,Waltham,MA,USA)according to the manufacturer's instructions.Then,RTmax II reverse transcriptase(Takara,Shiga,Japan)was used to reverse transcribe RNA.SYBR Green Master Mix(Thermo Fisher Scientific,Waltham,MA,USA)and StepOne Plus quantitative PCR system(Thermo Fisher Scientific,Waltham,MA,USA)were used to quantitatively analyze mRNA expression.Glyceraldehyde 3-phosphate dehydrogenase(GAPDH)was used as an internal control and to compare the differences in mRNA expression among the groups based on the 2-△△CTequation.All primer sequences are shown in Table S2.
2.7.Western blotting
Total protein was extracted using radioimmunoprecipitation assay lysis buffer(Beyotime,Shanghai,China).Quantitative analysis of protein concentration was performed using a BCA assay kit(Beyotime,Shanghai,China).After the samples were fractionated on an 8% polyacrylamide gel,the proteins were transferred to polyvinylidene difluoride membranes.Membranes were blocked with 5% bovine serum albumin diluted in Tris-buffered saline-Tween 20 buffer for 1 h at room temperature.The membranes were incubated with primary antibodies overnight at 4°C and then incubated with secondary antibody at room temperature for 1 h.The targeted protein bands were detected using a chemiluminescence reagent(Millipore,Burlington,MA,USA)and the density of bands was calculated using Image J.The dilutions of primary and secondary antibodies were as follows:TNC(1:1000,ZenBio,Chengdu,China),DNA methyltransferases(DNMT)1(1:1000,ZenBio,Chengdu,China),DNMT3a(1:1000,ZenBio,Chengdu,China),DNMT3b(1:1000,ZenBio,Chengdu,China),GAPDH(1:1000,ZenBio,Chengdu,China),and secondary antibody anti-rabbit IgG(1:2000,Cell Signaling Technology,Danvers,MA,USA).
2.8.Wound healing assay
After counting the NPC cells(cells for siRNA were initially treated for48 h)and adjusting the cell concentration to 1×105cells/mL,100μL of single cell suspension was seeded in 6-well plates and incubated until they were fully confluent.A straight wound was induced by scratching with a bacteria-free pipette and rinsing twice with phosphate-buffered saline.The gap was observed and photographs were taken under different microscopic fields at 0 and 24h.
2.9.Transwell assay
Transwell assay was performed using 24-well Millicell chambers containing Matrigel-coated membranes(Corning,NY,USA).After counting the NPC cells(cells for siRNA were initially treated for 48 h)and adjusting the cell concentration to 1×104cells/mL,100μL of single cell suspension in serum-free RPMI 1640 media was seeded in the upper chambers,and 15% fetal bovine serum diluted in RPMI 1640 media was placed in the lower chambers.After 24h,the cells stained with crystal violet were observed and photographed under different fields of the optical microscope.
2.10.Clonogenic survival assay
Cells were seeded in 6-well plates at densities of 2×102cells/well(0,2,and 4Gy)or 4×102cells/well(6 and 8 Gy).After 24h,the cells were irradiated with different X-ray doses.Seven days later,surviving colonies(≥50 cells/colony)were counted under the microscope after fixating and staining with Gentian violet.The surviving fractions(SFs)were calculated by dividing the plating efficiency(PE)by the PE of the non-irradiated control.Clonogenic survival curves were compared using GraphPad Prism.
2.11.Statistical analyses
All statistical analyses were performed using SPSS software(v 21.0)for Windows.Continuous data are presented as the mean±standard deviation(SD).A P value of<0.05 was considered statistically significant.
3.Results
3.1.Difference in DNA methylation profiles between CNE-2 and CNE-2R cells
First,we determined DNA methylation levels in CNE-2 and CNE-2R cells.A total of 1711 and 1679 methylation sites were found in CNE-2 and CNE-2R cells,respectively.Cluster analysis of the methylation sites identified in the sample showed that the genome-wide methylation levels of the two groups were different(Fig.1A).Compared to CNE-2 cells,the methylation status of 294 genes was significantly upregulated(PeakDMvalue≥0.2,PeakScore(-log10(P value)≥2)and that of 595 genes was significantly downregulated(PeakDMvalue ≥0.2,PeakScore≥2)in the CNE-2R cells(Fig.1B).Approximately 10 pathways were enriched in the KEGG pathway analysis(P<0.05,Fig.1C).In the analysis of GO functions,190 biological processes,24 cellular components,and 51 molecular functions were identified(P<0.05),and we showed the top 10 for each term(ranked by enrichment score,Fig.1D).Among these terms,the differential genes were mostly involved in single-multicellular organism processes,protein activation cascade,extracellular space,and channel activity,among others.

Fig.1.Difference in DNA methylation profiles between CNE-2 cell and CNE-2R cells.(A)Hierarchical clustering heatmap showing DNA methylation status in the three groups.(B)In the volcano plot,the two vertical lines are the PeakDMvalue=0.2 change boundaries,and the horizontal line is the statistical significance boundary(PeakScore-log10(P value)≥2).Compared with the CNE-2 group,items in the CNE-2R group with statistical significance as well as hypermethylation are presented as red dots,and hypomethylation is presented as blue dots.(C)KEGG pathway analysis of the differentially expressed genes showing the top 10 categories(ranked by enrichment score).(D)Corresponding gene enrichment using DAVID analysis showing the top 10 items for each term(ranked using enrichment score).SMY:Shengmai Yin;GO:gene ontology.
3.2.Changes in DNA methylation of CNE-2R cells after SMY intervention
After SMY intervention,1697 methylation sites were found in CNE-2R cells,and cluster analysis of the methylation sites identified in the sample showed that the DNA methylation status of CNE-2R cells had changed(Fig.1A).Compared to CNE-2cells,the methylation status of 172 genes was significantly upregulated(PeakDM-value≥0.2,PeakScore≥2),and the methylation status of 405 genes was significantly downregulated(PeakDMvalue≥0.2,PeakScore≥2)in the CNE-2R cells(Fig.2A).Our comparative analysis showed that the methylation status of 235 genes significantly upregulated and the methylation status of 361 genes significantly downregulated were restored to non-significant levels in CNE-2R cells after SMY intervention(Fig.2B).We performed GO enrichment analysis(P<0.05)and KEGG pathway analysis(P<0.05)on the genes that were restored to non-significant levels after SMY intervention.As shown in Fig.2C,the differential genes were mostly enriched for cellular component terms,including plasma membrane,extracellular region,extracellular space,and integral component of the plasma membrane.

Fig.2.Changes in DNA methylation of the CNE-2R cells after SMY intervention.(A)In the volcano plot,compared with the CNE-2 group,items in CNE-2R+SMY group with statistical significance as well as hypermethylation are presented as red dots,whereas hypomethylation is presented as blue dots.(B)Venn diagram of the differential genes among the CNE-2,CNE-2R,and CNE-2R+SMY groups.(C)Corresponding gene enrichment using DAVID analysis showing the top 5 items for each term(ranked by gene count).
3.3.TNC was downregulated and restored to partially
hypermethylated in CNE-2R cells after SMY intervention
TNC,which was included in the genes restored to nonsignificant levels after SMY intervention,and the GO term“extracellular region”are always overexpressed in cancer.To investigate the expression of TNC between CNE-2cells and CNE-2R cells we analyzed TNC gene expression from GSE48501 using GEO2R(GSE48501,gene expression data of human NPC cells and its radioresistant strain),which clearly showed that it was overexpressed in radioresistant cells.Therefore,TNC has the potential as a biomarker for early diagnosis(Fig.3A).We then confirmed the overexpression of TNC in CNE-2R cells and found through RT-qPCR and Western blotting that SMY intervention downregulated its expression(Figs.3B-D).

Fig.3.TNC was downregulated in CNE-2R cells after SMY intervention.(A)Tenascin-C(TNC)mRNA expression from GSE48501 analyzed by GEO2R.R:radioresistant;S:radiosensitive.(B)TNC mRNA expression(compared with the CNE-2R group,*P<0.05),as determined by real-time(RT)-qPCR.Each test was done at least thrice.(C and D)TNC,DNA methyltransferase(DNMT)1,DNMT3a,and DNMT3b protein expression(compared with the CNE-2R group,*P<0.05)were determined by Western blot analysis.Each test was done at least thrice.GAPDH:glyceraldehyde 3-phosphate dehydrogenase.
The differential expression of TNC may be mediated by its DNA methylation status.We visualized the methylation peak of TNC found in the chip using the NimbleGen SignalMap™data browser and found that methylation peaks increased in the CNE-2 and CNE-2R+SMY groups compared to the CNE-2R group(Fig.4A).As predicted by MethPrimer,we found a CpG island in the TNC promoter region(Fig.5).Methylation status was assayed by methylationspecific PCR(MSP),which showed that the TNC promoter was not methylated in the CNE-2R group,but was methylated in the CNE-2 and CNE-2R+SMY groups(Fig.4B).Bisulfate sequencing PCR(BSP)confirmed the result of MSP and we observed that TNC was restored to partial hypermethylation in CNE-2R group after SMY intervention,especially in the 5th CG site(Fig.4C).Furthermore,to investigate which methyltransferases were responsible for DNA methylation at CpG sites of the TNC gene,we determined the expressions of DNMT1,DNMT3a,and DNMT3b in three groups using Western blotting.As shown in Figs.3C and D,DNMT3a showed the lowest expression levels,whereas TNC exhibited higher expression levels and hypomethylation in CNE-2R cells.DNMT3a in CNE-2R cells was restored to the same expression level as that of CNE-2 cells after SMY intervention,suggesting that DNMT3a was most likely involved in the TNC gene methylation affected by SMY.

Fig.4.TNC was restored to partial hypermethylation in CNE-2R cells after SMY intervention.(A)TNC methylation peak in the chip visualized using NimbleGen's SignalMapTMdata browser.The X-axis represents location and the Y-axis represents peak score.(B)Inter-group comparison of DNA methylation status of TNC promoter region by methylation-specific PCR(MSP).M:methylated,U:unmethylated.(C)Inter-group comparison of DNA methylation status of TNC promoter region by bisulfate sequencing PCR(BSP).
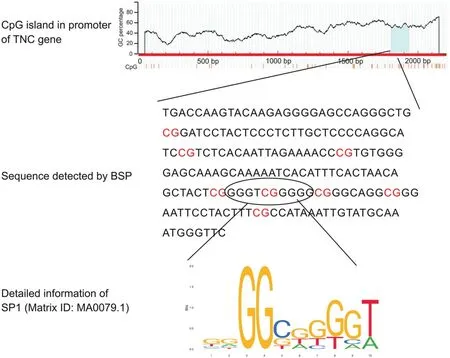
Fig.5.Potential specificity protein 1(SP1)binding sites in the promotor region of the Tenascin-C(TNC)gene.
3.4.Downregulation of TNC inhibited NPC cell radioresistance,migration,and invasion
The downregulation of TNC in CNE-2R cells after siRNA transfection was confirmed using RT-PCR and Western blotting,and after comparing with the control,we chose siRNA3 for further experiments(Fig.6A).
A survival clone test is a common method to measure the radiosensitivity of cells,as it reveals the survival score of irradiated cells.In a multi-target single-hit model,N represents the cell repair capacity of radiation lesions,with a larger N indicating a stronger cell resistance.The quasi-threshold dose(Dq)also has a direct ratio to cell resistance,whereas a larger mean lethal dose(D0)indicates stronger radioresistance.In this test,we counted surviving colonies(≥50 cells/colony)and calculated SFs for multi-target single-hit model matching(the correlation parameters of radiation biology are shown in Table 1).As shown in Table 1,the downregulation of TNC expression enhanced radiosensitivity in CNE-2R cells.

Table 1The correlation parameters of radiation biology of each group.
TNC is always overexpressed in cancer and is accompanied by cell proliferation and migration.The influence of TNC on radiosensitivity may be mediated by its effects on this mechanism.The wound healing assay revealed that CNE-2R cells with downregulated TNC expression healed significantly slower than the control cells over 24h.This indicated that downregulation of TNC inhibited NPC cell migration(Fig.6B).Moreover,the Transwell analysis showed that the downregulation of TNC expression was related to the significant inhibition of NPC cancer cell invasion(Fig.6C).

Fig.6.Downregulation of TNC inhibited nasopharyngeal carcinoma cell radiation resistance,migration,and invasion.(A)Expression of TNC(compared with the CNE-2R group,**P<0.01)was detected in CNE-2R cells by TNC and Western blotting after transfection with small interfering RNA(siRNA).(B)Representative image(left)of wound healing assay in each group.The area of cell migration was determined for statistical analysis(right)(**P<0.01).(C)Representative image(left)and histogram statistics(right)of the Transwell cell invasion assay,× 200 magnification(**P<0.01).NC:negative control.
4.Discussion
DNA methylation is receiving increasing attention as it is believed to play a role in different types of diseases.During the initiation and progression of cancer,epigenetic processes change,including the DNA methylation pattern[22].We investigated the correlation between DNA methylation and radiosensitivity of NPC cells in this study to further understand the heterogeneity in radiosensitivity between individuals.
As shown by the results,there was a significant difference in the genome-wide methylation status between CNE-2cells and its radioresistant strain.Compared to CNE-2cells,the methylation status of 294 genes was significantly upregulated and that of 595 genes was significantly downregulated in CNE-2R cells,indicating an overall genome-wide hypomethylation.SMY was found to restorethe methylation status of some of these genes,which means that SMY-mediated enhancement of radiosensitivity may be mediated by the restoration of its original DNA methylation status.We then carried out a GO enrichment analysis on the genes that showed restoration of methylation to non-significant levels after SMY intervention.Results showed that these genes were mostly enriched for cellular components,including the plasma membrane and extracellular region.In cancer,alteration of cellular plasma membrane homeostasis[23]and extracellular region components[24]may cause changes in multiple cellular processes,such as protein expression,endocytosis,apoptosis,cell proliferation,and metastasis.Moreover,in the results of the MeDIP array,we found differences in DNA methylation levels in some tumor suppressors,such as TP53 and PTEN,to be inconspicuous among the three groups,especially after SMY intervention.As is known,the mechanism of SMY radiosensitization is not simplified because of the complexity of SMY.Therefore,some ingredients may inactivate tumor suppressors,but others may work as demethylating reagents.At the same time,many TCMs have been reported to be used as demethylating reagents[25].
TNC,included in the genes restored to non-significant levels after SMY intervention,and the GO term “extracellular region”,are ECM glycoproteins that are always overexpressed in cancer,accompanying cell proliferation and migration[26].Recently,TNC has been confirmed to affect the expressions of important genes related to stem cell maintenance,such as the downregulation of dickkopf and inhibitor of differentiation of the two.Thus,we speculate that TNC may play a role in cancer stem cells[27].Accumulating evidence indicates that radioresistant cancer cells,which exhibit stem cell characteristics,are the main cause of radiation resistance[4].As TNC,which plays a role in the maintenance of tumor-stem cell stemness,has an effect on radiosensitivity.Here,we used Gene Expression Omnibus data,RTPCR,and Western blotting to investigate and compare the expression of TNC in CNE-2cells and its radioresistant strain,and the results showed that TNC was overexpressed in CNE-2R cells.This suggests that TNC may have potential as a biomarker.We found that SMY downregulated TNC expression and speculated that these changes may be mediated by the DNA methylation status of TNC,a hypothesis which we later confirmed.TNC was hypomethylated in CNE-2R cells,which was then partially restored to a hypermethylated state after SMY intervention,especially in the 5th CG site.The interactions between transcription factors and DNA are modulated by DNA methylation at CpG islands.Unmethylated CpG sites in promoters can activate genes by binding to transcription factors.In contrast,hypermethylated CpG sites cannot provide a binding platform for a complement of transcription factors.Specificity protein 1(SP1)typically binds unmethylated DNA to promote gene transcription,whereas binding to methylated CpG sites leads to inhibition,and is thus correlated with transcriptional silencing[28,29].We predicted potential SP1 binding sites in the promoter region of the TNC gene(http://jaspar.genereg.net/)and found that one of the SP1 binding sites was the 5th CG site that we had checked(Fig.5).Therefore,we speculate that the unmethylated CpG sites in CNE-2R cells provide a binding platform for SP1 to activate transcription.
Finally,we detected the expression of DNA methyltransferases(DNMT1,DNMT3a,and DNMT3b)and speculated that DNMT3a,which catalyzes de novo DNA methylation and prefers CpG sites in cells[30],was most likely involved in methylation in the TNC gene affected by SMY.In addition,after we knocked down DNMT3a by siRNA(sequence:CGAAGUGAAGGAGGAGAAC,Fig.S1),the qPCR result showed that compared with the CNE-2R group,there was no statistically significant difference in the expression of TNC mRNA in the CNE-2R+SMY group,which indicated that the expression of TNC mRNA increased.Considering the effects of DNMT3a,this result suggested that DNMT3a was the key protein in the DNA methylation of TNC.
Declaration of competing interest
The authors declare that there are no conflicts of interest.
Acknowledgments
We sincerely thank all of our colleagues for their generous support.This work was supported by the National Natural Science Foundation of China(Grant Nos.:81673718 and 82074132)and the Technology Project of Guangdong Province (Grant No.:2016A020226034).
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jpha.2020.11.010.
 Journal of Pharmaceutical Analysis2021年6期
Journal of Pharmaceutical Analysis2021年6期
- Journal of Pharmaceutical Analysis的其它文章
- Evaluation of the gastrointestinal anti-motility effect of Anacardium occidentale stem bark extract:A mechanistic study of antidiarrheal activity
- Simultaneous enantioseparation and simulation studies of atenolol,metoprolol and propranolol on Chiralpak®IG column using supercritical fluid chromatography
- Spectroscopic studies of the interaction between phosphorus heterocycles and cytochrome P450
- Impaired tricarboxylic acid cycle flux and mitochondrial aerobic respiration during isoproterenol induced myocardial ischemia is rescued by bilobalide
- Synergistic effects of methyl 2-cyano-3,11-dioxo-18beta-olean-1,-12-dien-30-oate and erlotinib on erlotinib-resistant non-small cell lung cancer cells
- A living cell-based fluorescent reporter for high-throughput screening of anti-tumor drugs