
Development and validation of a tumor microenvironment-related prognostic signature in lung adenocarcinoma and immune infiltration analysis*
2022-01-18 07:07:36ZhouLiYanqiFengPiaoLiShennanWangRuichaoLiShuXia
Zhou Li,Yanqi Feng,Piao Li,Shennan Wang,Ruichao Li,Shu Xia (✉)
1 Department of Oncology,Tongji Hospital,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430030,China
2 Department of Geriatric,Tongji Hospital,Tongji Medical College,Huazhong University of Science and Technology,Wuhan 430030,China
Abstract Objective Tumor-infiltrating immune cells and stromal cells in the tumor microenvironment (TME) significantly affect the prognosis of and immune response to lung adenocarcinoma (LUAD).In this study,we aimed to develop a novel TME-related prognostic model based on immune and stromal genes in LUAD.Methods LUAD data from the TCGA database were used as the training cohort,and three Gene Expression Omnibus (GEO) datasets were used as the testing cohort.The Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data algorithm was used to analyze the immune and stromal genes involved in the TME.Kaplan-Meier and Cox regression analyses were used to identify prognostic genes and construct a TME-related prognostic model.Gene set enrichment analysis and TIMER were used to analyze the immune features and signaling pathways of the model.Results A TME-related prognostic model based on six hub genes was generated that significantly stratified patients into the high-and low-risk groups in terms of overall survival.The model had strong predictive ability in both the training (TCGA) and testing (GEO) datasets and could serve as an independent prognostic factor for LUAD.Moreover,the low-risk group was characterized by greater immune cell infiltration and antitumor immune activity than the high-risk group.Importantly,the signature was closely associated with immune checkpoint molecules,which may serve as a predictor of patient response to immunotherapy.Finally,the hub genes BTK,CD28,INHA,PIK3CG,TLR4,and VEGFD were considered novel prognostic biomarkers for LUAD and were significantly correlated with immune cells.Conclusion The TME-related prognostic model could effectively predict the prognosis and reflect the TME status of LUAD.These six hub genes provided novel insights into the development of new therapeutic strategies.
Key words:lung adenocarcinoma;tumor microenvironment;immunotherapy;immune checkpoint molecules;prognostic biomarkers
Lung cancer is the leading cause of cancer-related deaths worldwide,with a predicted 5-year survival rate of 16%[1].More than 85% of cases are classified as non-small cell lung cancer (NSCLC),with lung adenocarcinoma (LUAD) being the most common pathological subtype[2].In recent decades,the discovery of driver gene mutations in tumors has allowed for the introduction of personalized molecular-targeted therapy for NSCLC[3].However,this approach is not feasible for treating tumors that do not carry gene alterations,and the inevitable resistance to tyrosine kinase inhibitors further suggests the need for alternative therapeutic options in lung cancer patients[4].In recent decades,immunotherapy targeting immune checkpoints has made great progress in the treatment of NSCLC[5].Immune checkpoint inhibitors (ICIs) enhance the antitumor effects of the immune system to obtain a potent and durable cure[6].However,the overall response rate to ICIs is relatively low,and only one-fifth of cancer patients benefit from these agents[7].Therefore,it is necessary to identify novel biomarkers for predicting LUAD patient survival and response to ICI therapy.
A growing body of evidence has demonstrated the importance of the tumor microenvironment (TME) in oncogenesis and tumors[8].The TME is a complex network composed of tumor cells,immune cells,mesenchymal stem cells,fibroblast cells,endothelial cells,inflammatory mediators,and extracellular matrix[9].The interactions between tumor cells and their surrounding supporting cells contribute to the malignant biological behaviors of cancer,such as unlimited proliferation,resistance to apoptosis,and evasion of immune surveillance[10].Therefore,the TME significantly affects the therapeutic response to and clinical outcomes of patients with cancer.The major non-tumor components of the TME,tumor-infiltrating immune cells and stromal cells have been proposed to be valuable for the diagnostic and prognostic assessment of patients with tumors[11,12].The development of a comprehensive model of the TME based on immune and stromal signature genes may contribute to the prognostic evaluation of LUAD patients and predict the efficacy of immunotherapy.With advancements in sequencing techniques,bioinformatics tools such as Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data (ESTIMATE) and CIBERSORT,make it feasible to estimate the distribution of immune and stromal cells in the TME by analyzing specific gene expression signatures of immune and stromal cells[13].This algorithm has been successfully applied to quantitative analysis of the TME in various tumors and the identification of immune and stromal genes involved in the TME,and its effectiveness has been proven[14].
To date,several predictive models have been constructed for LUAD prognosis stratification,mainly focusing on immune-related genes or immune cells[15].However,few studies have investigated the influence of TME on LUAD patient survival outcomes and response to immunotherapy,specifically based on immune and stromal components.To fill this knowledge gap,we aimed to develop a TME prognostic model based on immune and stromal genes to predict the survival outcomes and immune responses.In the present study,we systematically investigated the expression details and clinical significance of immune and stromal genes in the TME of LUAD and developed a novel TME-related prognostic model.In addition,we validated this model using independent datasets and analyzed its potential prognostic mechanism and association with immunotherapy responses.Our findings provide promising biomarkers for the prognostic stratification and selection of patients responsive to ICIs,which would facilitate accurate management and appropriate personalized therapies for patients with LUAD.
Materials and methods
Data source and preprocessing
The gene expression profiles of 594 LUAD case were downloaded from the TCGA database (https://portal.gdc.cancer.gov/),along with their corresponding clinical and survival data.Datasets GSE26939,GSE37745,and GSE29016,which contained microarray expression data and clinical information of LUAD patients,were downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/).In our study,data from TCGA were used as the training cohort,whereas the three GEO datasets were used as validation cohorts.
Generation of the immune score and stromal score
ESTIMATE is an algorithm for estimating the infiltration of immune and stromal cells in tumor samples by analyzing the specific gene expression signatures of immune and stromal cells.Here,we calculated the immune and stromal scores to predict the proportion of immune and stromal components in each sample using the ESTIMATE algorithm with the aid of the R software estimate package.
Identification of differentially expressed genes related to the TME and functional enrichment analysis
All patients in the training cohort were divided into high and low immune/stromal score groups according to the median immune and stromal scores,respectively.Kaplan-Meier analysis was conducted to compare the survival difference between the two groups,and the p-value of the log-rank test was calculated.The limma package was used to identify differentially expressed genes (DEGs) between the high and low immune/stromal score groups with a fold change (FC)=1 and false discovery rate (FDR) < 0.05.DEGs between the high and low immune score groups were defined as immune DEGs,whereas the DEGs between the high and low stromal score groups were defined as stromal DEGs.Finally,the intersecting genes between the immune and stromal DEGs were considered for subsequent analysis.Heatmaps of DEGs were generated to show expression differences using the heatmap package heatmap.Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses of intersecting DEGs were performed using the clusterProfiler,enrichplot,and ggplot2 packages.Only terms with p-and q-values of < 0.05 were considered significantly enriched.Moreover,we downloaded a list of immune-related genes from the Immunology Database and Analysis Portal (Immport) to select immune-related DEGs among these DEGs using the VennDiagram package.
Construction and evaluation of the TME-related prognostic model in the training set
Univariate Cox and Kaplan-Meier analyses were performed in the training cohort to identify significant prognostic genes among the immune-related DEGs.APvalue < 0.05 in the log-rank test was considered significant.Multivariate Cox regression analysis was performed to obtain the respective coefficients (βi) of each gene.Finally,the TME prognostic model was constructed on the basis of the key prognostic genes,and the risk score of each patient was calculated on the basis of the expression level of each key prognostic gene and its regression coefficient.
Kaplan-Meier and receiver operating characteristic (ROC) analyses were used to assess the accuracy of the model in predicting clinical outcomes.Univariate and multivariate Cox regression analyses were performed to evaluate the prognostic value of the model and other common clinical factors such as age,sex,stage,and TNM stage.
Validation of the TME-related prognostic model in the testing set
The feasibility and stability of the TME-related prognostic model were confirmed using the GEO database model.Patients in the three testing datasets were divided into the high-and low-risk groups according to the formula of risk score derived from the training dataset using the same methods as above.Kaplan-Meier survival analysis and ROC curve analysis were used to evaluate the performance of the six-gene prognostic model in predicting the outcomes of patients with LUAD.
Evaluation of immune status between the high-risk and low-risk groups stratified by prognostic model
To explore the potential mechanism of the prognostic effects of the model,we analyzed the immune status and pathway enrichment of high-risk and low-risk samples.First,we quantified the enrichment levels of the 29 immune signatures in each LUAD sample using singlesample gene set enrichment analysis (ssGSEA) score.Based on the ssGSEA score,we performed a hierarchical clustering analysis to compare immune activities between the high-risk and low-risk samples.CIBERSORT is an algorithm used for estimating the proportion of immune cell subsets through cell type identification by estimating the relative subsets of RNA transcripts.In this study,we used the CIBERSORT algorithm to construct 21 types of immune cell profiles in LUAD samples and compared the differences in immune cell subtypes between the high-and low-risk groups.KEGG enrichment analysis was performed to analyze the functions or pathways that were upregulated in the two groups.Finally,we compared the mRNA levels of immune checkpoints and their ligands and the expression of HLA genes in the high-and lowrisk groups.
Comprehensive analysis of prognostic hub genes in the model
To reveal the regulatory mechanisms of the prognostic hub genes in the TME,we systematically analyzed the genetic alterations and functional enrichment of these six genes.First,RNA expression and gene-encoding protein expression level alterations in LUAD compared with normal tissue were estimated by the Wilcoxon test and immunohistochemistry (IHC),respectively.IHC results for hub genes were obtained from the Human Protein Atlas (HPA) database.The String online database and Cytoscape software were used to construct a proteinprotein interaction (PPI) network between the molecules.We then analyzed the pathways of hub genes by gene set enrichment analysis (GSEA),using the gene expression level as the phenotype.The curated KEGG gene set was downloaded from the Molecular Signature Database,and FDR < 0.05 was considered significant.Finally,we evaluated the correlation between hub gene expression and immune cell infiltration in LUAD using TIMER.
Results
Immune scores and stromal scores were correlated with survival outcomes
A total of 510 LUAD cases from TCGA were used as the training cohort,and three GEO datasets were used as the validation cohorts.The clinical information for all cohorts is summarized in Table 1.We calculated the immune and stromal scores of each LUAD patient in TCGA and divided them into high and low immune/stromal score groups on the basis of the median value.Kaplan-Meier survival analysis showed that patients with high immune and stromal scores showed better survival outcomes than those with low scores,with log-rank tests ofP=0.01 and 0.026,respectively (Fig.1a,1b).
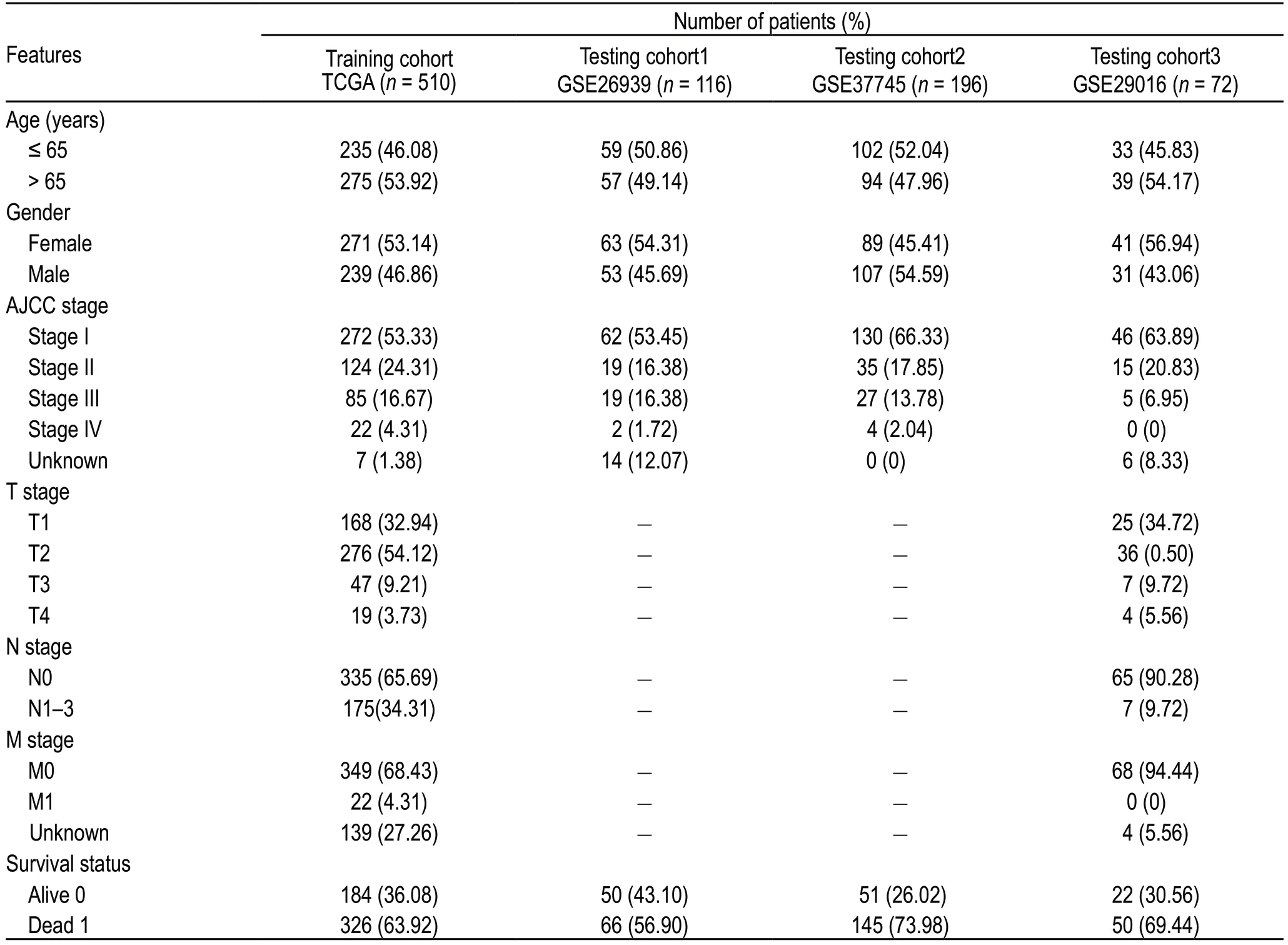
Table 1 Clinical characteristics of LUAD patients included in this study
Identification of DEGs based on immune score and stromal score
The heatmap showed that genes in the high score group had lower expression levels than those in the low score group,both for immune and stromal scores (Figure 1C,D).A total of 776 immune DEGs were obtained from the comparison of immune scores (samples with high scores vs.low scores),of which 613 genes were upregulated and 163 genes were downregulated.Similarly,792 stromal DEGs were obtained from a comparison of the stromal scores,consisting of 678 upregulated genes and 114 downregulated genes.Moreover,Venn diagrams showed that 297 DEGs were commonly upregulated in the high-score groups,and 66 DEGs were commonly downregulated (Fig.1e,1f).These notable DEGs were potentially determinant factors of TME status.
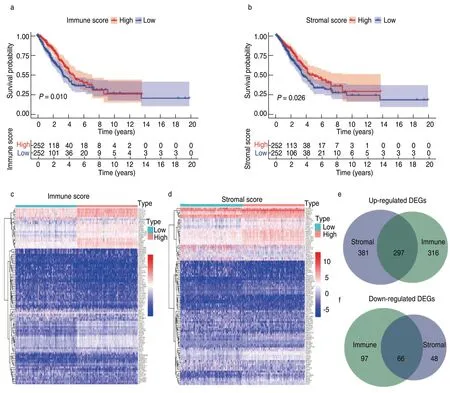
Fig.1 Identification of differential expressed genes(DEGs).(a) Kaplan-Meier survival curve of high and low immune score groups;(b) Kaplan-Meier survival analysis for high and low stromal score groups;(c) Heatmap for DEGs generated by comparison of gene expression profiles in high and low immune score groups;(d) Heatmap for DEGs in high and low stromal score groups;(e,f) Venn diagrams showed the common up-regulated and downregulated DEGs shared by immune and stromal score groups
GO and KEGG enrichment analysis
Results of the GO enrichment analysis showed that these DEGs were mainly involved in immune-related functions,such as T-cell activation and lymphocyte proliferation (Fig.2a,2c).KEGG analysis also revealed enrichment of the T cell receptor signaling pathway,chemokine signaling pathway,and hematopoietic cell lineage (Fig.2b,2d).Since the DEGs were correlated with immune functions or pathways in LUAD,we further identified the top 89 immune-related DEGs from the Immport database for subsequent analysis (Fig.3a).
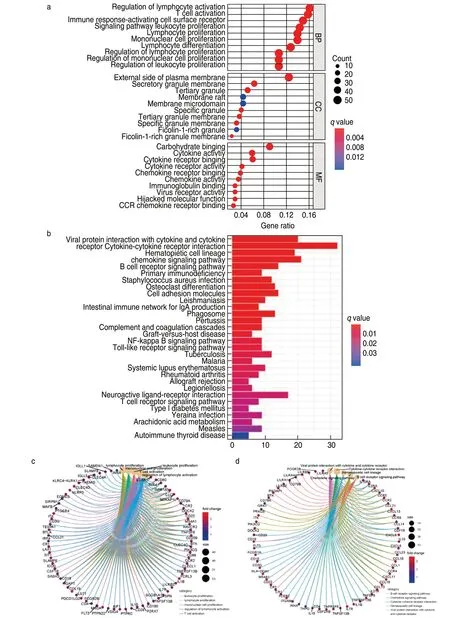
Fig.2 Functional enrichment analysis.(a) GO enrichment analysis of 363 DEGs;(b) KEGG enrichment analysis of DEGs;(c)Circle plot show the DEGs involved in top 5 enriched terms of GO analysis;(d)Circle plot show the DEGs involved in top 5 enriched terms of KEGG analysis
Construction of the TME-related prognostic model
Univariate Cox and Kaplan-Meier analyses were conducted to determine the significant prognostic genes among the 89 immune-related DEGs.A total of 24 genes were identified as significant in the Cox regression analysis (Fig.3b),of which 6 genes were also significant in the Kaplan-Meier analysis.Among them,higher expression levels of BTK,CD28,PIK3CG,TLR4,and VEGFD correlated positively with poor survival outcomes,whereas INHA expression correlated negatively with prognosis (Fig.3c).Then,the six prognostic genes were subjected to multivariate Cox regression analysis,and the risk coefficient of each gene was calculated (Table 2).The TME-related prognostic model was constructed as follows:risk score=(-0.111958) × EXPBTK+0.279096 × EXPCD28+0.008079 × EXPINHA+(-0.357674) × EXPPIK3CG+0.099561 × EXPTLR4+(-0.102261) × EXPVEGFD.
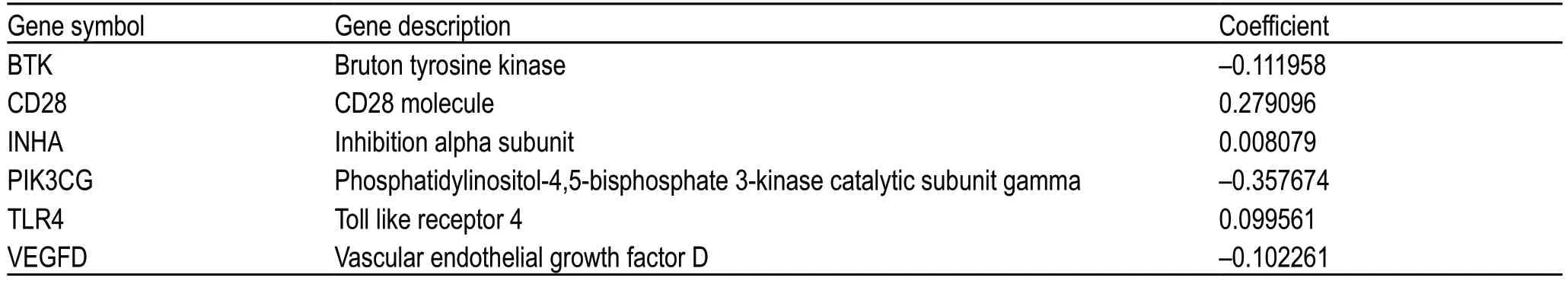
Table 2 Genes in the TME-related prognostic model
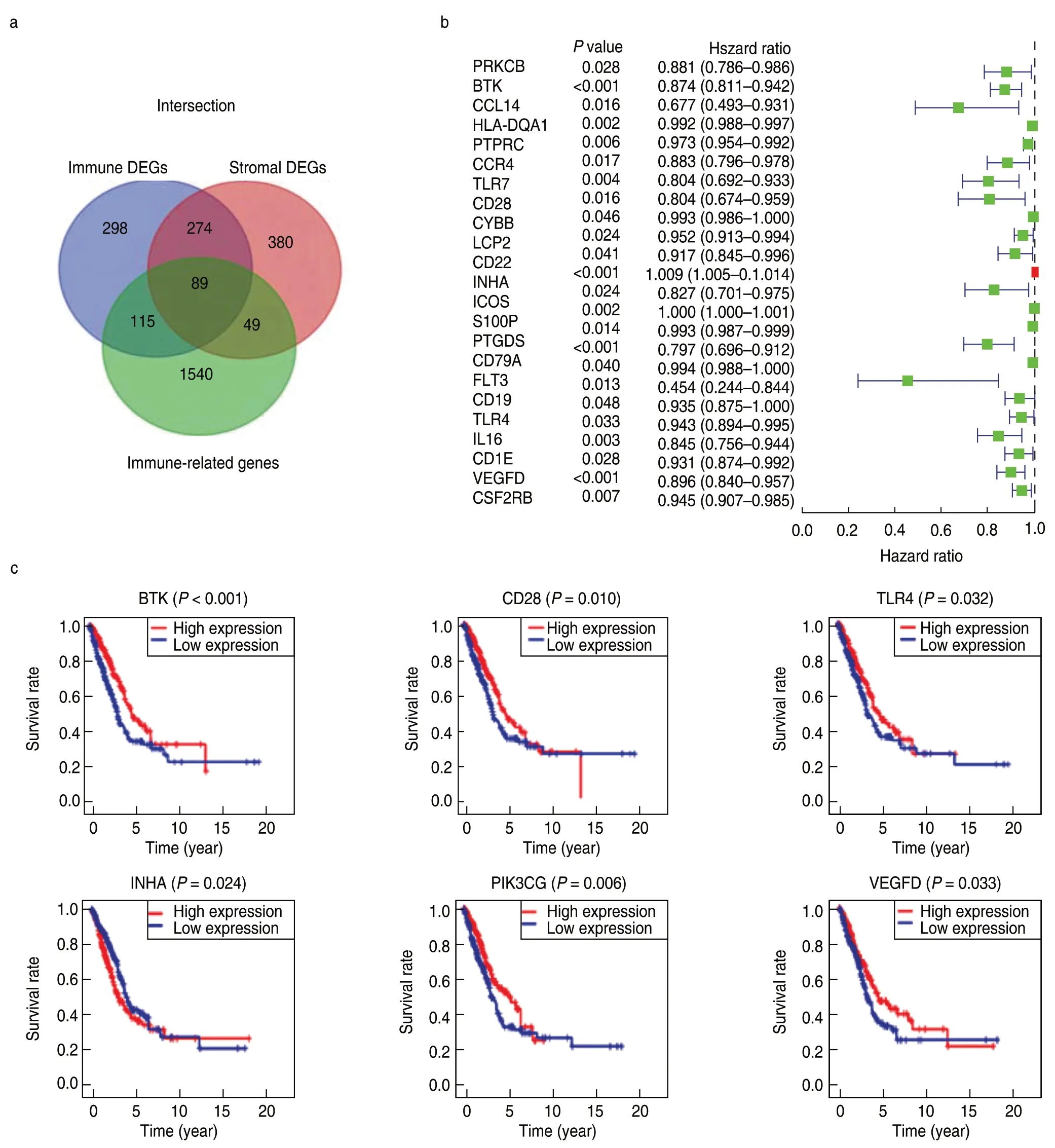
Fig.3 Univariate Cox and Kaplan Meier analysis for prognostic genes screening.(a) Identification of immune-related DEGs;(b) The forest plot of 24 prognostic immune-related DEGs screened out by Univariate Cox regression analysis with P < 0.005;(c) Survival curves of the 6 prognostic genes extracted by the Kaplan-Meier analysis.Patients were labeled with high expression or low expression according to the median expression level of the 6 genes
Prognostic value of the TME-related model in the training and validation cohorts
We calculated the risk score for each patient in the training cohort (n=510) and divided them into the high-and low-risk groups according to the median cutoff value (cutoff value:-0.261).The Kaplan-Meier plot showed that patients in the high-risk group had worse survival outcomes than those in the low-risk group (Fig.4a).The ROC curve of the 5-year survival prediction was drawn to assess the predictive accuracy,with an area under the curve of 0.688 (Fig.4b).Additionally,the risk curve indicated that the high-risk group had a higher mortality and worse prognosis than the low-risk group (Fig.4c,4d).The prognostic value of our model in patients with LUAD was further evaluated using other common prognostic factors.Although univariate Cox analysis indicated that the pathological stage and risk score had prognostic effects,only the risk score could be used as an independent prognostic factor (P< 0.001;Fig.4e,4f).
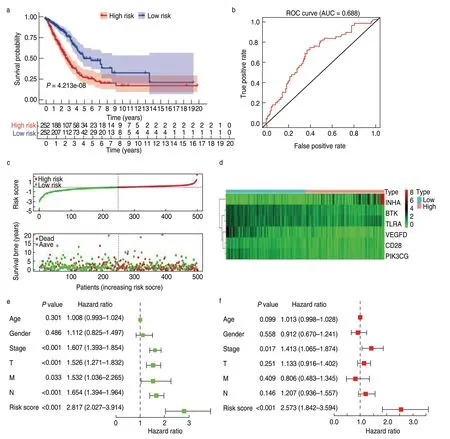
Fig.4 Construction and validation of the TME-related prognostic model in the training cohort.(a) Kaplan-Meier survival curve of low-and high-risk groups stratified by the TME-related prognostic model;(b) The ROC analysis of the TCGA dataset for survival prediction by the TME prognostic model;(c) The distribution of risk score and survival time in high-and low-risk groups;(d) Heatmap of the six prognostic genes;(e)The Univariate Cox analysis evaluating the prognostic effect of the model and common clinical factors;(f) Multivariate Cox analysis evaluating independent predictive ability of our model for overall survival
Consistent with the results in the training dataset,the six-gene model stratified the samples of the three GEO testing datasets into high-risk and low-risk groups.Patients with low-risk scores had better survival outcomes than those in the high-risk group (P< 0.05;Fig.5a-5c).The areas under the ROC curves for predicting 5-year survival in the three testing datasets were 0.679,0.666,and 0.732 (Fig.5d-5f).These results suggest that the TME-related prognostic model is robust in predicting the survival outcomes of patients with LUAD.
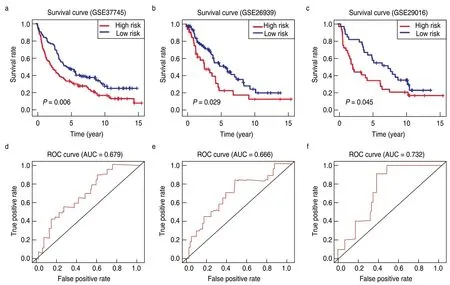
Fig.5 Validation of the TME-related prognostic model in the testing cohort.Kaplan-Meier survival curves showing overall survival outcomes of high-and low-risk groups in GSE37745 (a),GSE26939 (b) and GSE29016 (c).The ROC curves for judging the predictive accuracy of the model in GSE37745 (d),GSE26939 (e) and GSE29016 (f)
Evaluation of the immune status between low-risk and high-risk groups
The strong stratification power of the TME-related model in predicting the survival of patients with LUAD led us to explore the difference in functional characteristics between the two risk groups.The ssGSEA score of the 29 immune signatures was used to evaluate the immune status of the two groups.The heatmap showed that the low-risk group had a higher immune activity than the high-risk group (Fig.6a).Consistent with the ssGSEA results,immune and stromal scores in the low-risk group were significantly higher,and the tumor purity of the low-risk group was significantly lower (Fig.6b-6d).This finding indicated that more immune and stromal cells infiltrated the TME of low-risk samples,whereas more tumor cells were present in high-risk samples.Moreover,we identified the immune cell subtypes in the two groups.The low-risk group had a significantly higher proportion of memory B cells,memory CD4+T cells,monocytes,and dendritic cells than the high-risk group,whereas the high-risk group had a markedly higher proportion of M0 macrophages (Fig.6e).Taken together,these results suggest that patients with low-risk scores show elevated antitumor immune activity,leading to more favorable clinical outcomes.
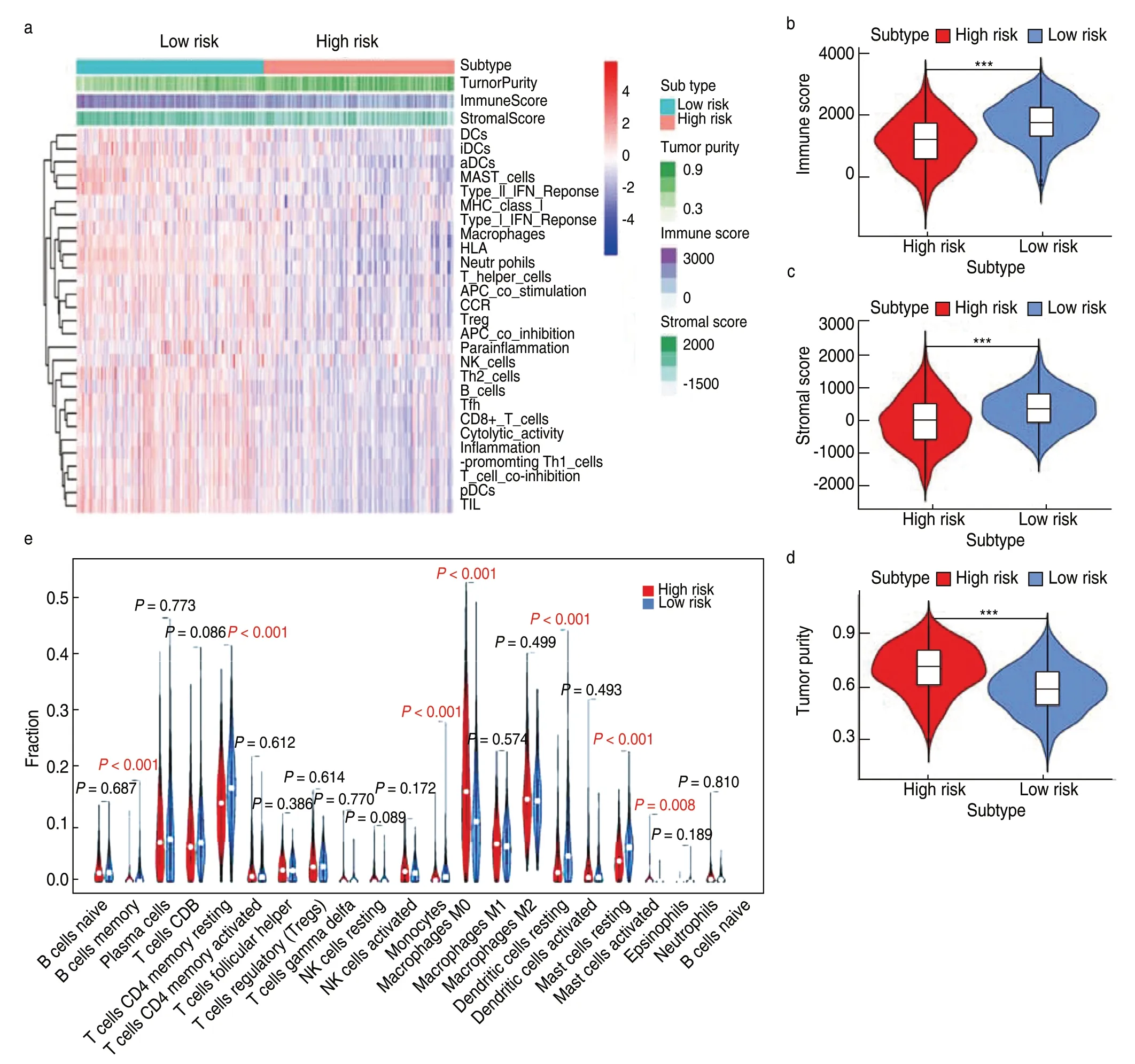
Fig.6 Evaluation of immune status and immune cell infiltration levels between high-and low-risk groups.(a) The heatmap of the overall immune status of high-and low-risk groups in TCGA database,showing greater heterogeneity between the two groups;(b-d) The violin plots showed the difference in immune score,stromal score and tumor purity between low-and high-risk groups.***P < 0.001;**P < 0.01;*P < 0.05;(e) The violin plot shows the difference in the proportion of 21 kinds of immune cells between high-and low-risk groups,and the Wilcox rank-sum was used for the significance test
Potential mechanisms of the prognostic effects of the TME-related model
GSEA was conducted to elucidate the specific regulatory mechanisms resulting in the differences in prognosis and immune status between the two risk cohorts.The results showed that the low-risk group was enriched not only in immunoregulation and immune cell activation,but also in many cancer-associated pathways,such as JAK-STAT signaling,cell adhesion molecules,and transendothelial migration (Fig.7a).In contrast,the high-risk group was impoverished in immune signatures but enriched in metabolic signaling.Notably,most HLA genes showed significantly higher expression in the lowrisk group than in the high-risk group,indicating that local immune regulation and immunogenicity were more active in the low-risk group (Fig.7b).
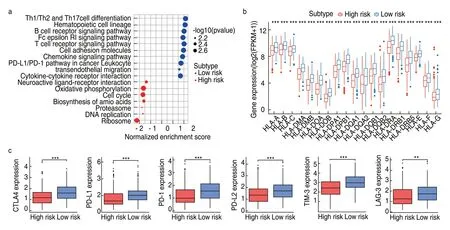
Fig.7 Functional mechanisms of the TME-related model and association with immune checkpoint molecules.(a) KEGG pathways enriched in high-and low-risk samples;(b) The expression profiles of HLA genes of low-and high-risk groups.***P < 0.001;**P < 0.01;*P < 0.05;(c) Comparison of expression levels of CTLA-4,PD-1,PD-L1,PD-L2,TIM-3 and LAG-3 between high-risk and low-risk groups (Wilcox rank-sum test)
Relationship between the TME-related model and immunotherapy response
In recent years,immune checkpoint proteins such as cytotoxic T lymphocyte antigen 4 (CTLA-4) or the programmed cell death ligand 1/protein 1 pathway (PD-L1/PD-1) have been used as crucial targets for immunotherapy in LUAD[16].We explored the relationship between the model and immunotherapy response by analyzing the expression of common immune checkpoints in the high-and low-risk groups and found that the expression of CTLA-4,PD-1,PD-L1,PD-L2,TIM-3,and LAG-3 in the low-risk group was significantly higher than that in the high-risk group (allP< 0.001;Fig.7c).This suggests that patients with low-risk scores might respond better to ICI treatment than those with high-risk scores because the expression of immune checkpoint molecules tends to be positively associated with immunotherapeutic responsiveness.Therefore,the construction of a risk cohort using our model could be a good stratification method for patients with LUAD regarding whether to conduct immunotherapy.
The mechanism of action of prognostic hub genes in the TME
To further analyze the potential function of the hub genes,our results were verified using the HPA and TIMER databases.We found that CD28 and INHA had significantly higher expression levels in LUAD samples than in normal lung samples,whereas BTK,PIK3CG,TLR4,and VEGFD had lower expression levels in tumor tissues (Fig.8a).In terms of protein levels,the protein expression patterns of BTK,INHA,PIK3CG,TLR4,and VEGFD were consistent with their RNA-seq expression alterations (Fig.8b).However,CD28 showed no significant difference.The PPI network also showed extensive interactions among BTK,CD28,PIK3CG,and TLR4.
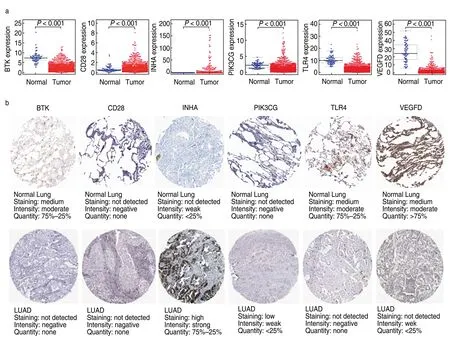
Fig.8 Expression profiles of the six hub genes in the model.(a) The expression levels of the six prognostic genes in LUAD samples and normal lung samples in the TCGA database (BTK,CD28,INHA,PIK3CG,TLR4 and VEGFD).Wilcox test was used to calculate the significance level between the two groups;(b) The immunohistochemistry results reflecting the gene-encoding protein levels of the six hub genes in LUAD and normal lung tissues from the HPA database
GSEA suggested that BTK,CD28,PIK3CG,TLR4,and VEGFD were enriched in the same pathways.High expression of these five genes was mainly correlated with immune-related activities,such as antigen processing and presentation,the chemokine signaling pathway,and the JAK-STAT signaling pathway,whereas low expression of these genes was associated with metabolic pathways (Fig.9a,9b).In contrast,high expression of INHA was correlated with metabolic pathways,and low expression was involved in the activation of immune pathways (Fig.9c,9d).More importantly,the expression of BTK,CD28,PIK3CG,TLR4,and VEGFD positively correlated with the infiltration of CD4+T cells,CD8+T cells,B cells,neutrophils,dendritic cells,and macrophages (Fig.10).However,INHA was negatively correlated with infiltration of the six immune cells.Collectively,these results suggest that these six hub genes affect the immune activity of the TME.

Fig.9 The GSEA enrichment analysis of the hub prognostic genes.(a) The enrichment pathways of high expression of BTK,CD28,PIK3CG,TLR4 and VEGFD.Each line representing one particular pathway with unique color,only pathways with p and q < 0.05 were considered significant.And only several leading gene sets were displayed in the plot;(b) The enrichment pathways of low expression of the five genes;(c) Enrichment pathways of high INHA expression;(d) Enrichment pathways of the low INHA expression
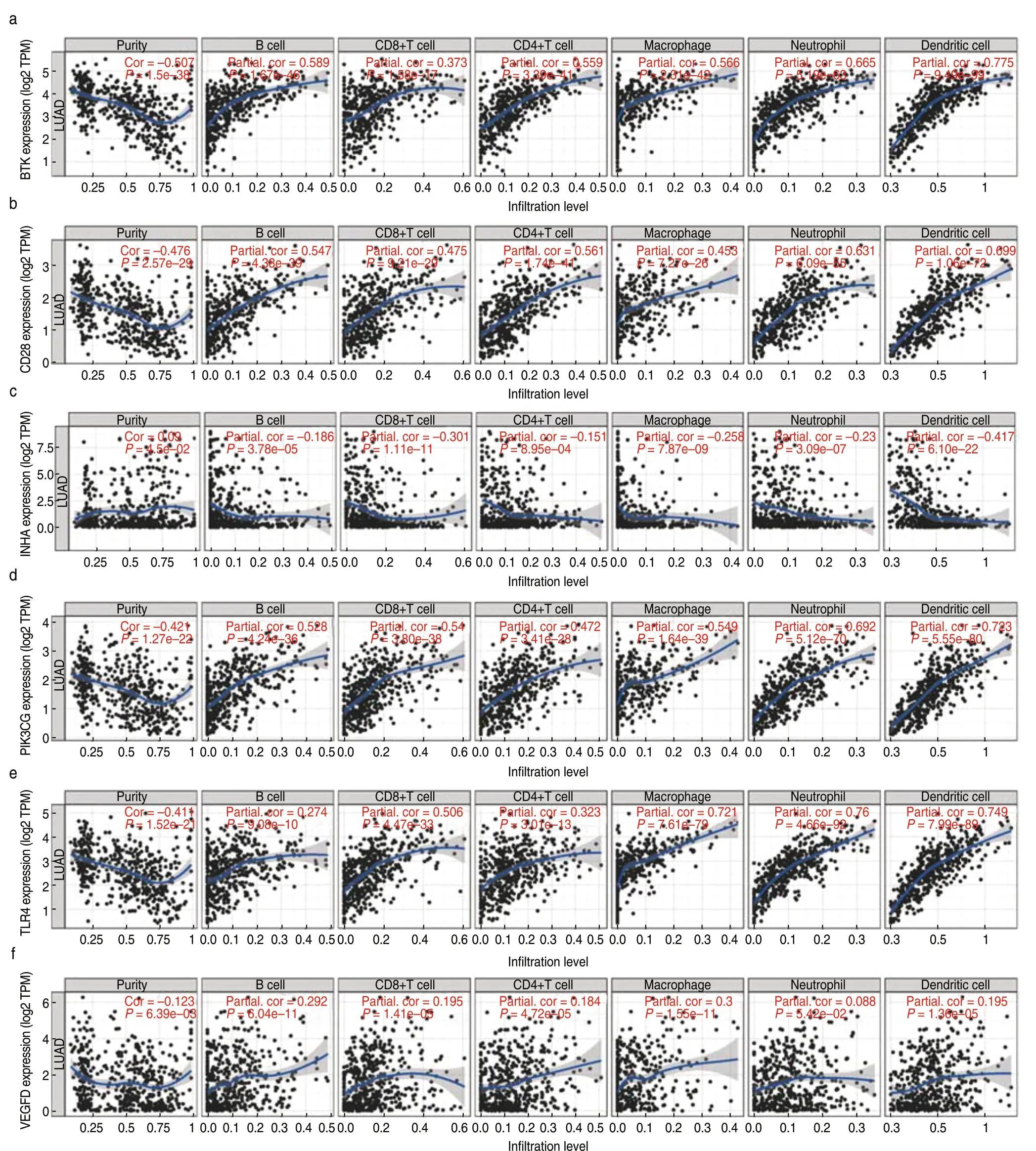
Fig.10 Correlation between hub prognostic genes and immune cell infiltration.(a-f) The gene expression levels against tumor purity are displayed on the left-most panel
Discussion
In this study,we aimed to identify a novel TMErelated prognostic model for LUAD.We embarked on TME-related DEGs generated by comparing the immune and stromal scores in LUAD samples.Subsequently,a list of TME-related genes that contribute to the survival outcomes of patients with LUAD was extracted.Finally,a six-gene prognostic model,based on prognostic TMErelated genes,was constructed.Both immune and stromal genes in LUAD samples were analyzed to better reflect the complete TME status.Furthermore,we validated its prognostic value in three testing sets from the GEO datasets.Kaplan-Meier and ROC analyses revealed the strong predictive ability of our model for LUAD prognosis in both the training and testing sets.Univariate and multivariate Cox analyses confirmed the independent prognostic value of the six-gene model.Accordingly,unlike those developed previously,the TME-related prognostic model developed herein could reflect the tumor immune microenvironment status and predict the prognosis of LAUD more accurately.Moreover,an enhanced understanding of the model and related hub genes would help to elucidate regulatory mechanisms of the TME and develop new treatment strategies.
Experimental and clinical studies have demonstrated that the immune and stromal components of the TME play significant roles in lung cancer development and progression[17].The immune and stromal cells infiltrating the TME are composed of different cell types.As the most important immune cells in the TME,tumor-infiltrating T lymphocytes execute key effector cytotoxic functions and mediate responses to ICIs[18].Tumor-associated macrophages are another class of immune cells that interact with lung cancer cells.Macrophage-tumor cell interactions lead to the release of pro-inflammatory cytokines,chemokines,and growth factors,which in turn recruit additional inflammatory cells to the microenvironment[19].Other immune cells in the TME include B cells,NK cells,dendritic cells (DCs),T regulatory cells (Tregs),and B regulatory cells (Bregs).Cancer-associated fibroblasts are the most abundant stromal cells in the TME and play critical roles in the inflammatory response and immune suppression of tumors[20].Fibroblasts promote tumor progression via multiple pathways,including regulation of the extracellular matrix,production of growth factors or cytokines,and promotion of angiogenesis,whereas some fibroblast subtypes also show antitumor activities by secreting immunosuppressive cytokines[21].
To determine the distinct gene expression profiles in the TME with respect to immune and stromal components,DEGs based on immune and stromal scores were screened.Six prognostic hub genes among these DEGs were identified (survival positive correlation:INHA;negative correlation:BTK,CD28,PIK3CG,TLR4,and VEGFD).Interestingly,functional analysis showed that BTK,CD28,PIK3CG,TLR4,and VEGFD could promote immune infiltration,while INHA inhibited immune cell infiltration.These results suggest that the six hub genes participate in the immune regulation of the TME and affect the prognosis of patients with LUAD,which might be potential immune prognostic markers and therapeutic targets for LUAD.
Among these six genes,Bruton tyrosine kinase (BTK) is a member of the Tec kinase family.As a key component of the B-cell antigen receptor signaling pathway,BTK plays a vital role in B lymphocyte development,differentiation,and signaling[22].Ibrutinib is a small-molecule irreversible inhibitor of BTK that has been approved for the treatment of hematological malignancies and some solid tumors owing to its ability to inhibit tumor growth by modifying the tumor microenvironment and its potential synergistic activity with ICIs[23,24].CD28 is a key T-cell costimulatory molecule that binds to B7 molecules,which are involved in the regulation of T cell proliferation and activation,along with cytokine production[25].Previous studies have demonstrated that CD28 can predict the response to anti-PD-1 therapy in patients with lung cancer[26].PIK3CG encodes the PI3Kγ enzyme,which can activate the signaling molecule Akt and modulate various cell functions such as cell proliferation,migration,and adhesion[27].Novel PI3K inhibitors are important for the treatment of hematologic malignancies[28].The protein encoded by TLR4 is a member of the toll-like receptor family.Studies have shown that TLR4 is highly expressed in immune cells,such as monocytes and lymphocytes,but is expressed at low levels in epithelial,endothelial,and cancer cells.Thus,TLR4 agonists have been widely explored as potential immunotherapeutic agents for the treatment of cancer[29].VEGFD belongs to the vascular endothelial growth factor family and can induce both angiogenesis and lymphangiogenesis[30].Clinical studies have shown that low expression of VEGFD is a predictor of greater survival benefits from bevacizumab treatment in patients with CRC[31].INHA encodes a member of the transforming growth factor-beta (TGF-beta) superfamily of proteins.However,its function in lung cancer remains unknown.Our results suggest that high expression of INHA is associated with a poor prognosis of LUAD.A possible mechanism might be that INHA is involved in vascularization and tumor metastasis,leading to a poor prognosis[32].Further studies are required to clarify the role of these hub genes in the TME in the initiation and development of LUAD.
Finally,a TME-related prognostic model was developed using the six hub immune genes for survival prediction.The low-risk group showed higher expression of HLA genes.HLA-related genes play a significant role in immune regulation,and their expression is advantageous for immunotherapy efficacy[33].Our results showed elevated antitumor immune activity in the lowrisk group,which could explain why the low-risk group had more favorable clinical outcomes than the high-risk group.In addition to survival prediction,this TME-related signature was also a predictor of patient response to ICI treatment.To date,many biomarkers have been verified to indicate the efficacy of ICI treatment,including TMB,PD-L1 expression level,neoantigens,and gut microbiota[34].Generally,most biomarkers reflect the status of the tumor immune microenvironment in a certain aspect.Thus,a prognostic model based on the TME may aid in the stratification of patients with LUAD to identify those responsive to immunotherapy.It is possible that patients with low-risk scores are more sensitive to immunotherapy than those with high-risk scores,since immune checkpoint molecules are more highly expressed in low-risk groups,and the increased levels of immune checkpoints indirectly indicate pre-existing T cell activation in the low-risk group.
This study has some limitations.The six-gene model was derived from retrospective data,and more prospective data are needed to validate our results.Second,this study lacked basic experiments to validate the function of the six hub genes and their association with immune cell infiltration.Third,patients receiving immunotherapy were not included in this study;therefore,the predictive ability of the model for immunotherapy response was evaluated indirectly.
Conclusions
In conclusion,we constructed a TME-related prognostic model to predict LUAD patient survival outcomes and responses to immunotherapy.Patients with low risk scores had better prognoses and were expected to benefit from ICI treatment.This model might be valuable for prognostic management and patient selection before immunotherapy and deserves further validation.A significant association was observed between the hub genes and patient prognosis and immune infiltration,providing novel insights for the development of new treatment strategies.
Acknowledgments
We are grateful to the TCGA database and GEO database for the source of data used in our study.
Conflicts of interest
The authors indicated no potential conflicts of interest.
 Oncology and Translational Medicine2021年6期
Oncology and Translational Medicine2021年6期
- Oncology and Translational Medicine的其它文章
- Autophagy-related lncRNA and its related mechanism in colon adenocarcinoma
- Effect of UBR5 on the tumor microenvironment and its related mechanisms in cancer*
- GFPT2 pan-cancer analysis and its prognostic and tumor microenvironment associations*
- Malnutrition as a predictor of prolonged length of hospital stay in patients with gynecologic malignancy:A comparative analysis*
- Effect of radiotherapy on tumor markers and serum immune-associated cells in patients with esophageal cancer*
- Correlation analysis of breast fibroadenoma and the intestinal flora based on 16S rRNA sequencing*