
节段性皮肤僵硬综合征一例
2022-01-18 08:39:06陈声利卢宪梅陈学超
中国麻风皮肤病杂志 2022年3期
陈声利 卢宪梅 刘 红 陈学超
山东第一医科大学附属皮肤病医院(山东省皮肤病医院),山东省皮肤病性病防治研究所,济南,250022
临床资料患者,男,5岁。左股部皮肤硬化4年余。患者4个月时左股部皮肤发硬,逐渐扩展,曾于当地医院就诊,考虑硬皮病、嗜酸性筋膜炎、结缔组织痣,未治疗。患儿父母非近亲结婚,足月剖腹产出生,出生体重3.5 kg。家族中无类似患者,否认遗传病史。体格检查:患儿一般情况良好,营养、发育及智力均正常。心肺腹部查体未见异常。左膝关节活动不受限。皮肤科检查:左股部自腹股沟至膝上方皮肤弥漫性硬化,与下方组织粘连,不能捏起,与周围组织分界不清,左股比右侧略细,皮损表面毛发增多(图1)。实验室检查:血尿常规、肝肾功能未见异常。皮损组织病理示:表皮轻度乳头瘤状增生,真皮胶原纤维增粗,排列紊乱,胶原纤维间可见脂肪细胞,未见明显炎症细胞浸润(图2)。阿新蓝染色:真皮内黏蛋白增多(图3)。基因检测未发现既往报道致病基因FBN1的突变。诊断:节段性皮肤僵硬综合征。建议患者积极进行功能锻炼,目前仍在随访中。
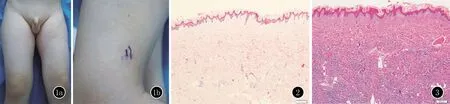
图1 1a: 左股部皮肤硬化,比右腿略细;1b:毛发增多 图2 表皮轻度乳头瘤状增生,真皮中深部胶原纤维增粗,排列紊乱,胶原纤维间见脂肪细胞(HE,×200) 图3 真皮内黏蛋白沉积(阿新蓝染色,×200)
讨论皮肤僵硬综合征(stiff skin syndrome,SSS),又称为先天性筋膜发育不良。目前国内外报道较少,检索PUBMED和知网,共查到约60例报道,临床易误诊。1971年,Esterly和Mckusick[1]首次描述皮肤僵硬综合征。国内由耿松梅于2002年首次报道[2]。SSS的病因和发病机制尚不清楚,目前认为由于fibrillin-1(FBN1)编码的基因突变,导致转化生长因子(TGF-β)信号活化,从而使成纤维细胞异常增殖[3]。目前所报道的存在FBN1基因突变的家系均符合常染色体显性遗传特征,且受累患者均属于泛发型SSS,节段性SSS未能找到其致病突变[4]。
SSS发病年龄较早,多发于婴儿期或儿童期。好发于臀部和股部。临床表现为皮肤硬化,伴有轻-中度毛发增多,进展缓慢,严重时关节活动受限。Myers等[5]对已报道的52例SSS进行了分析,认为SSS可分为节段性SSS和弥漫性SSS两种。节段性SSS以单侧皮损为主,平均发病年龄4.1岁,男女发病率为3.5∶1;弥漫性 SSS 累及双侧,平均发病年龄1.6岁,男女发病率相同,男性略多,97%的患者关节活动受限,而节段性SSS中有44%的患者关节活动受限。平均随访时间11年,报道的18例节段性SSS都没有进展为双侧受累。节段性SSS和弥漫性SSS的组织病理表现相同。SSS的病理学特征为:胶原束增厚,水平排列,缺乏炎症,胶原间黏蛋白增多。Myers等[5]认为在胶原间出现脂肪细胞是诊断该病的一个主要特征。
SSS应与硬皮病、硬肿病、嗜酸性筋膜炎、结缔组织痣鉴别。(1)硬皮病的皮损为象牙色或白色的致密硬化,无毛发增多的现象。组织病理学特征为真皮网状层胶原纤维束致密、均质化,在早期阶段,有密集的淋巴细胞、浆细胞浸润。晚期阶段,皮肤附属器萎缩消失。(2)硬肿病多见于成人,好发于颈项和背部。组织病理学特征是真皮网状层显著增厚,胶原间黏蛋白沉积。(3)嗜酸性筋膜炎起病迅速,临床特征是疼痛、触痛性硬结,表现为凹陷性水肿、橘皮样外观。病理表现为浅筋膜明显增厚、纤维化,真皮网状层深部可出现慢性炎细胞浸润。实验室检查外周血嗜酸粒细胞增多。(4)结缔组织痣表现为坚实的单个或多发性呈肤色的丘疹、结节或斑块,病理表现为增加的胶原束无规则排列。
本例患儿婴儿期发病,发生于一侧股部,临床表现为皮肤硬化,毛发增多。病理表现为真皮网状层胶原纤维增粗,无炎细胞浸润,黏蛋白增多,胶原纤维间可见大小不一的脂肪团块。基因检测未发现FBN1的突变。综合临床和病理表现可排除其他皮肤硬化性疾病,符合节段性SSS的诊断。尤其是病理特征中胶原纤维间可见大小不一的脂肪团块,与Myers等[5]对该病的病理特征的观点一致。
皮肤僵硬综合征病情进展缓慢,尚无有效治疗方法,故以对症治疗为主。应尽早进行康复理疗,以防止局部功能障碍。
猜你喜欢
中国皮肤性病学杂志(2023年11期)2023-10-30 06:14:26
当代医药论丛(2023年2期)2023-02-28 12:36:42
当代医药论丛(2022年20期)2022-11-17 02:13:24
中国生殖健康(2019年11期)2019-01-07 01:27:32
青少年科技博览(中学版)(2017年5期)2018-02-28 21:23:59
西南国防医药(2016年7期)2016-12-01 06:01:16
实用手外科杂志(2015年3期)2015-08-27 01:53:18
中国医疗美容(2015年2期)2015-07-19 10:11:59
西部皮革(2015年3期)2015-04-16 05:07:31
中华皮肤科杂志(2014年3期)2014-12-19 12:54:41
