
加权基因共表达网络分析筛选胰腺癌肿瘤免疫相关基因
2022-01-11 09:28李慧月王沐榛田雪梅
华南师范大学学报(自然科学版) 2021年6期
李慧月, 王沐榛, 喻 叶, 田雪梅
(华南师范大学生命科学学院, 广州 510631)
胰腺癌是人类最具侵袭性和致命性的恶性肿瘤之一,胰腺癌的早期诊断较为困难,大多数患者发现时已经是晚期. 手术切除、化疗在胰腺癌治疗中的效果十分有限[1]. 免疫治疗是近年来肿瘤治疗研究的热点,在非小细胞肺癌和黑色素瘤等实体瘤中展现了不错的临床疗效,但由于胰腺癌独特的肿瘤微环境和较低的肿瘤免疫原性,限制了免疫治疗对胰腺癌的疗效[2-4]. 肿瘤浸润性免疫细胞是肿瘤微环境的主要细胞成分,其组成和功能可随着宿主免疫状态的改变而改变,并与肿瘤细胞之间存在复杂的相互作用,影响肿瘤的进展和预后[5-7]. 全面揭示胰腺癌肿瘤浸润性免疫细胞的特征,了解胰腺癌的免疫表型对胰腺癌患者的早期诊断和治疗具有重要意义.
加权联合表达网络分析(Weighted Gene Co-expression Network Analysis, WGCNA)可用于探索疾病临床特征与基因簇之间的相关性,以确定癌症的相关模块和中心基因[8]. 该方法已被广泛用于在转录组水平寻找生物标志物[9]. 本研究使用反卷积CIBERSORT法计算样本的肿瘤浸润性免疫细胞的相对比例,结果显示B细胞所在的模块相关性最高,接着通过WGCNA识别出与B细胞相关的重要模块和枢纽基因,随后利用公共数据库进行一系列验证,最终分析筛选出胰腺癌中与B细胞免疫浸润相关的分子标志物.
1 材料和方法
1.1 数据来源和预处理
人胰腺癌样本的mRNA测序数据从TCGA数据库( https:∥portal.gdc.cancer.gov/)的“PAAD”队列下载得到,共获得182个样本数据,包括178例胰腺癌组织样本和4例正常胰腺样本. 同时下载临床资料数据(Clinical). 在R语言环境下运行“limma”包对基因表达数据进行归一化处理后用于后续分析.
1.2 肿瘤浸润性免疫细胞的评估
CIBERSORT反卷积法是基于已知参考集LM22,通过分析 mRNA 表达数据来定量评估每个样本中 22 种免疫细胞亚型的相对比例的分析方法[10],目前已广泛运用于肿瘤相关免疫细胞的研究中. 在本研究中,使用R包“CIBERSORT”,基于参考基因表达特征集“LM22”,将TCGA中胰腺癌的mRNA表达矩阵导入算法进行反卷积分析和比对,得到胰腺癌样本中22种免疫细胞的相对比例.
1.3 构建加权基因共表达网络和模块分析
加权基因共表达网络分析WGCNA是用来分析不同样本之间基因关联模式的系统生物学方法,可以用来鉴定高度协同变化的基因表达矩阵,并根据表达矩阵的内连性以及基因与表型之间的关联度鉴定候选核心基因. 本研究使用R包“WGCNA”鉴定相关基因模块,筛选可能与胰腺癌免疫相关的基因集. 通过分析不同加权系数下模块的尺度独立性和平均连通性来确定模块分析的软阈值β. 软阈值确定后,构建无尺度拓扑分布网络,根据基因间的Pearson相关系数将相关矩阵转换为邻接矩阵,进一步转换为拓扑重叠矩阵(Topological Overlap Matrix,TOM),得到基因间的相异度(1-TOM). 利用层次聚类函数,将表达谱相似的基因归类于同一个基因模块.
分析各模块的组成部分. 将模块与临床特征进行关联分析,本研究将22种免疫细胞作为临床特征纳入共表达网络,采用Pearson检验来计算各模块与免疫细胞浸润程度之间的相关性. 当P<0.05时,选取相关系数最高的模块定义为中心模块.
为了确定基因在中心模块中的功能,使用网页工具“Metascape”(http:∥metascape.org)用于通路富集分析[11],设置P<0.05为截止点.
1.4 枢纽基因的识别
通过WGCNA分析模块内连通性筛选前30个候选基因,显著性模块定义为P<0.05. 选取中心模块中的所有基因,使用检索工具String(https:∥string-db.org/)检索相互作用的基因[12],并构建PPI网络,设置置信度评分大于0.4为截止标准,以TSV格式导出. 然后将PPI网络导入到Cytoscape工具中进行可视化操作[13],利用CytoHubba插件的Degree评分来识别前30个中心节点. 使用网页工具Venny 2.1.0(https:∥bioinfogp.cnb.csic.es/tools/venny/)对WGCNA分析得到的30个候选基因和PPI分析得到的30个中心节点绘制韦恩图,取交集内所包含的基因作为本研究的枢纽基因.
TIMER(https:∥cistrome.shinyapps.io/timer/)是一个系统分析不同癌症类型的免疫浸润的综合资源[14],它通过反卷积的统计方法,从基因表达谱推断肿瘤浸润性免疫细胞的相对比例[15]. 使用TIMER来计算目标免疫细胞的浸润水平与枢纽基因表达之间的Spearman相关性.
1.5 差异表达基因和生存分析
原始数据在R语言环境(version3.5.3, https:∥www.r-project.org/)下使用“affy”包进行预处理和标准化[16]. 使用“limma”包筛选胰腺癌患者和对照组表达数据中的差异表达基因(DEGs),设定阈值:|log2FC|>2,P<0.05;采用Kaplan-Meier分析和logrank检验评估枢纽基因表达对胰腺癌患者总生存率的影响[17].
为了验证枢纽基因的表达方式,使用了来自癌症微阵列数据库Oncomine(https:∥www.oncomine.org)的4个独立的微阵列数据集进行meta分析[18]. Oncomine将大量公布的癌症微阵列转录组数据与先进的分析工具结合在一起,能够分析人类癌症中原癌和抑癌基因的通路、调控网络和功能网络.
1.6 统计分析
根据数据分布特征,采用非参数检验或t检验分析实验组和对照组之间差异的统计学意义. 所有R语言分析均用R3.5.3软件进行,P<0.05被认为有统计学意义.
2 结果
2.1 mRNA表达数据
本研究获得了来自癌症基因组图谱(The cancer genome atlas,TCGA)数据库的182个样本RNA seq数据,包括178例胰腺癌组织样本和4例正常胰腺样本. 将mRNA表达数据合并成一个行为基因名、列为样本名的标准化矩阵,得到一个包含19 669个基因、182个样本的表达矩阵. 为剔除样本中与其他基因联系不够密切的基因,简化运算,基于R环境下使用“apply”函数选取了变化量较大的(方差处于前四分之一)的4 916个基因进行进一步分析.
2.2 肿瘤浸润性免疫细胞的评估
CIBERSORT计算胰腺癌中的22种肿瘤浸润性免疫细胞成分,选择7种免疫细胞:B cells memory ,NK cells activated,Dendritic cells activated,Macrophages M1,Macrophages M2,Neutrophils,Monocytes作为WGCNA的临床特征数据,保存为文本格式用于后续分析.
2.3 加权基因共同表达网络的构建和模块分析
利用R包“WGCNA”构建4 916个基因表达值的共表达网络,计算平均连接值和Pearson相关值对样本进行聚类. 当以0.9作为相关系数时,软阈值β=12. 通过WGCNA分析,构建了13个共表达模块,每个模块至少包含30个基因. 以模块特征基因和免疫细胞的Pearson相关系数表示二者的相关性,相关性排名前3的模块分别为:记忆B细胞所在的cyan模块、树突状细胞所在的royalblue模块以及NK细胞所在的greenyellow模块,相关系数分别为0.52、0.45和0.37(图1),选择其中相关系数最高的记忆B细胞所在的cyan模块内的基因进行后续分析. 使用Metascape工具对该模块中的基因进行通路分析和过程富集分析(图2),“B细胞增殖”是第3个高度富集的条目,进一步说明了cyan模块和B细胞的高度相关性.
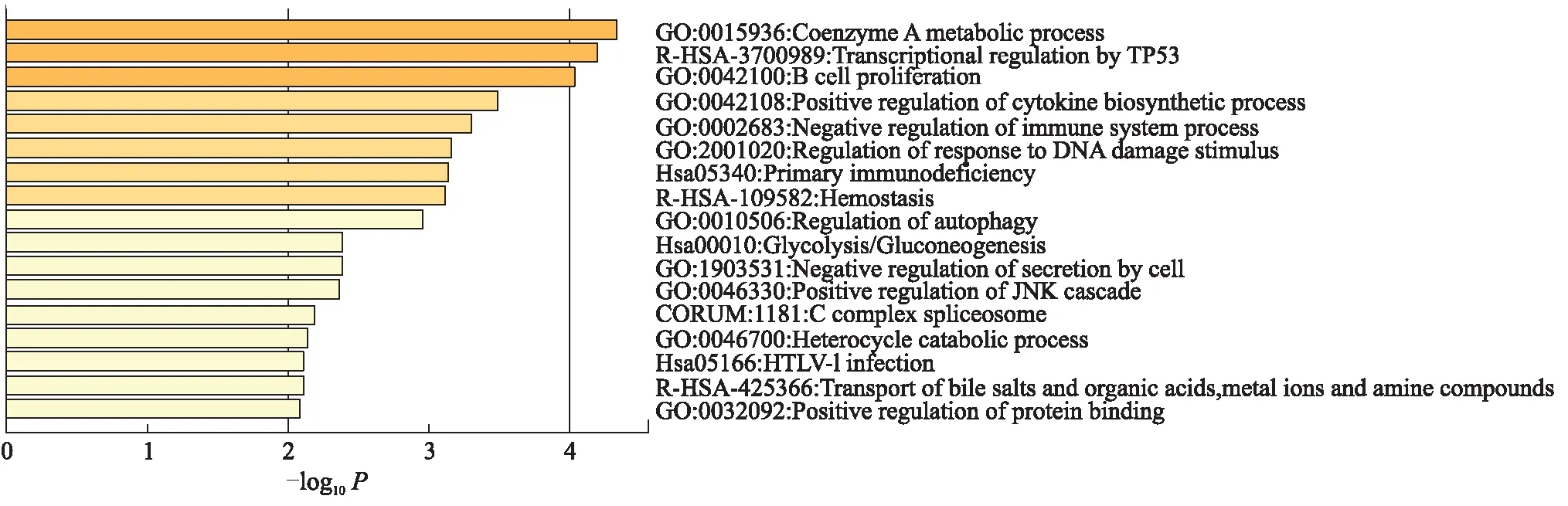
图2 cyan模块内基因富集的生物学过程条目
2.4 枢纽基因的识别
通过以下步骤筛选胰腺癌中与B细胞相关的枢纽基因. 首先,基于WGCNA分析在cyan模块中进行基因连通性排序,筛选出前30个候选基因;其次,在PPI网络中,运用Cytoscape的插件CytoHubba对网络中的基因进行连接度Degree值排序,得到Degree评分前30个节点作为中心节点,置信度评分大于0.4 (图3);最后,进行韦恩分析,得到候选基因与中心节点的交集,共筛选出9个枢纽基因:CD79B、MYC、BANK1、TIMELESS、CD19、ATF3、ITGAL、IKZF3、RRAGB.
为了研究这些枢纽基因与B细胞的关系,分析了这些基因在TIMER数据库中的表达数据,结果显示:9个基因的表达值与B细胞浸润水平呈正相关(除ATF3外,所有基因的P值均小于0.05)(图4).
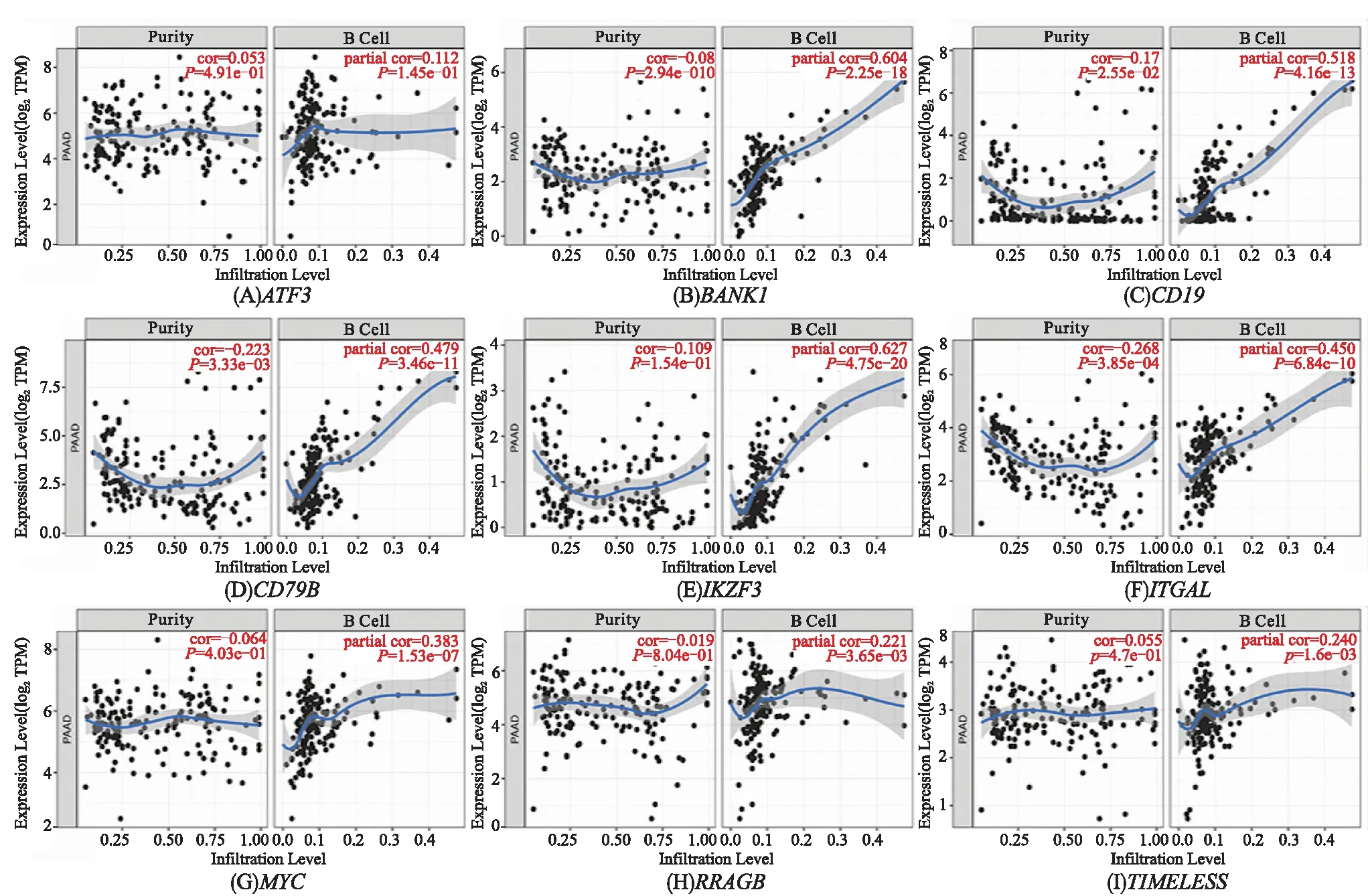
图4 枢纽基因与B细胞浸润程度的相关性
2.5 差异表达基因和生存分析
利用R语言的“limma”包对TCGA中的胰腺癌组织和正常组织的基因表达数据进行处理,得到了9个基因的表达水平,发现ITGAL在肿瘤组织中的表达水平明显高于正常对照组(|log2FC|>2,P<0.05)(图5). 采用Kaplan-Meier分析方法对9个枢纽基因进行分析,结果显示MYC、BANK1、ITGAL、RRAGB具有统计学意义(P<0.05)(图6). 对于MYC和BANK1,高表达患者生存预后较差,而ITGAL和RRAGB高表达的患者预后较好.结合差异表达基因和生存分析,选择ITGAL作为潜在的生物标志物进行进一步分析.
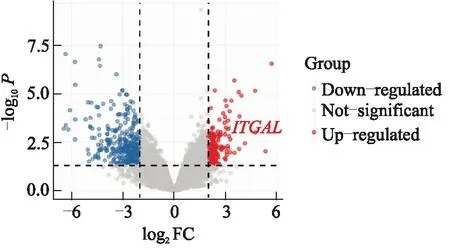
图5 差异基因表达的火山图
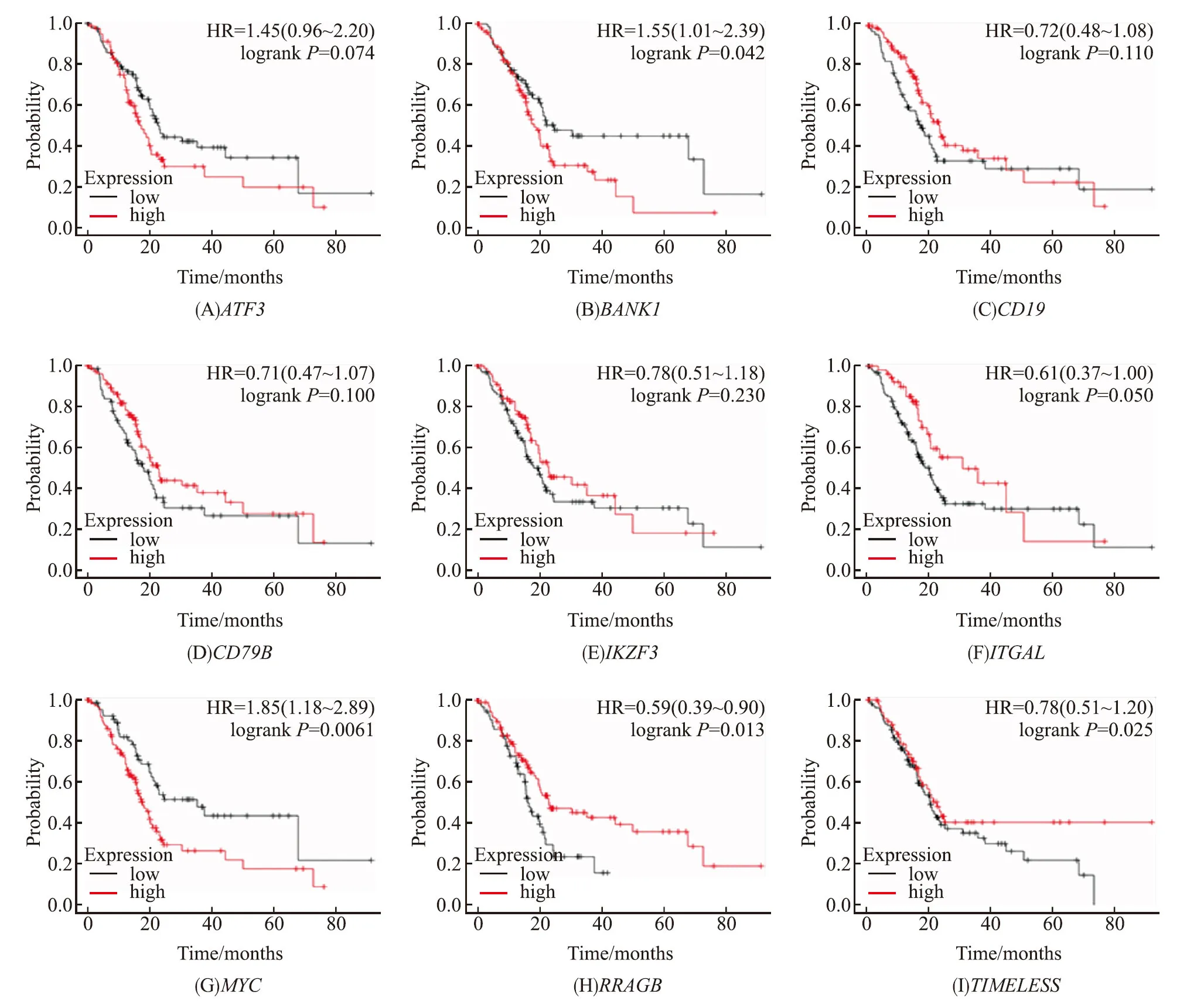
图6 枢纽基因的Kaplan-Meier曲线
为了验证ITGAL在肿瘤组织和正常组织中的差异表达,对Oncomine数据库中3个数据集的4个分析进行了meta分析. 结果显示:ITGAL在肿瘤组织中显著高表达,这与TCGA数据集的分析一致(Median Rank Values=1 809,P<0.05)(图7).

图7 基于Oncomine数据集的基因表达的meta分析
3 讨论
胰腺癌是全球最致命的恶性肿瘤之一,常规的治疗手段如手术、化疗和放疗效果不佳. 近年来,免疫治疗在一些恶性肿瘤中取得了很大的成功,不断有新的靶点被发现,免疫治疗也是人们不断探索的治疗胰腺癌的方法[19]. 尽管在癌症研究领域使用WCGNA已经发现了的一些关键生物标志物[20-21],但大多数研究局限使用差异表达基因来构建加权基因共表达网络,这可能导致一些在癌症过程中起重要作用的基因的缺失. 因此,本研究对TCGA中胰腺癌数据进行严格的质量控制和评估后,将表达数据中的4 916个基因全部纳入WCGNA分析,共分为13个模块. 接下来,本研究发现了与B细胞相关性最高的模块. 此外,近期的研究表明,B细胞在免疫治疗中发挥着非常重要的作用. JONSSON等[22]发现三级淋巴结构中的B细胞与T淋巴细胞协同作用,最终可能靶向肿瘤细胞. 经过免疫检查点抑制剂治疗后,有应答者的B细胞受体的多样性高于无应答者,提示B细胞可能具有更好的识别肿瘤抗原的能力[23]. 因此,本研究将B细胞浸润水平作为临床信息. 结合基因表达数据和临床信息,鉴定出9个与B细胞浸润水平相关的关键模块的枢纽基因,提示了这些基因参与胰腺癌进展的潜在机制. 在TIMER数据库中分析这9个基因与B细胞的关系,均显示出显著的正相关. 随后进行差异表达基因分析和生存分析,结果显示ITGAL表达最为显著.ITGAL在肿瘤组织中高表达,当它在胰腺癌组织中高表达时,有着更好的预后.
ITGAL编码淋巴细胞功能相关抗原-1 (LFA-1)的α亚基,表达于淋巴细胞表面[24]. 一方面,LFA-1主要与内皮细胞上的胞内黏附分子-1 (ICAM-1)相互作用,介导B细胞活化和产生免疫球蛋白,通过促进B细胞黏附来降低B细胞活化的阈值[25-26]. ICAM-1在肿瘤组织中的过表达,与肿瘤侵袭转移、免疫功能调节有关. 胰腺癌组织中ICAM-1的表达明显高于正常组织,提示ICAM-1与胰腺癌的进展的相关性,ICAM-1可能作为胰腺癌早期诊断的生物标志[27-28]. 因此,ITGAL作为与ICAM-1相互作用的分子有可能是胰腺癌免疫治疗的潜在靶点.
猜你喜欢
现代仪器与医疗(2022年4期)2022-10-08
保健医苑(2022年6期)2022-07-08
抗癌(2021年4期)2021-12-01
中学生数理化(高中版.高一使用)(2021年2期)2021-03-19
房地产导刊(2020年8期)2020-09-11
家庭影院技术(2019年4期)2019-04-17
领导决策信息(2018年16期)2018-09-27
当代陕西(2018年12期)2018-08-04
当代陕西(2018年12期)2018-08-04
数学学习与研究(2017年3期)2017-03-09
