
α-四氢萘酮肟酸酯及其衍生物的合成研究
2022-01-08 02:38詹庆明徐永琴杨维清
四川轻化工大学学报(自然科学版) 2021年6期
詹庆明,刘 瑶,李 柳,徐永琴,杨维清
(西华大学理学院,成都 610039)
引 言
肟及其衍生物是一种很有价值的合成原材料[1]。它们可以通过贝克曼重排反应制备中间体或脱水反应制备其他种类重要的中间体[2-3]。另外酮肟酸酯与烯基硼酸、炔或者烯烃的过渡金属催化偶联反应成为合成取代吡啶的主要方法[3],其中酮肟酸盐的N-O断裂作为氧化剂,可以使氧化还原反应在温和的条件下进行[4-6]。肟与芳基卤化物或芳基硼酸的偶联已被用于合成有重要应用价值的邻芳基羟胺。芳香肟还可以与α-重氮羰基化合物在过渡金属催化体系下通过分子间C-H键功能化环化合成多种异取代喹啉N-氧化物[7-11]。
肟的主要合成方法为醛结构的化合物与盐酸羟胺在合适的碱性条件下反应制备得到:(1)醛溶解到相应的溶剂当中,加入盐酸羟胺和三乙胺,室温下搅拌反应数小时制备得到肟产物[12],该方法由于盐酸羟胺的溶解度问题,导致产物的产率偏低,反应时间较长;(2)醛溶解到水中与盐酸羟胺进行反应[13-15],该方法也因为盐酸羟胺溶解不足,导致产物产率偏低。而肟酸酯的主要合成方法有:(1)肟在弱酸强碱盐的条件下与乙酸酐反应制备得到相应的肟酸酯结构[16-17],由于乙酸酐的环保性差与危害性,使得该方法的应用比较困难;(2)肟与卤代酸酐反应制备得到肟酸酯[18-20],但是卤代酸酐是一种较为危险的化合物,操作相对比较困难;(3)肟结构与乙酸反应制备得到肟酸酯[21-22],该方法反应条件相对比较苛刻,目标产物产率较低。
因此,通过对前人合成肟酸酯的方法进行总结,研究一种快速、高效的合成α-四氢酮肟及其衍生物和α-四氢酮肟酸酯及其衍生物的方法。
1 实验部分
1.1 实验试剂和仪器
盐酸羟胺、甲醇(MeOH)、乙醇(Et OH)、氢氧化钠(NaOH)、乙酸钠(AcONa)、二氯甲烷(DCM)、乙酸乙酯(EA)与4-二甲氨基吡啶(DMAP)均为分析纯,购自成都科龙化学试剂有限公司;α-四氢萘酮、7-溴-3,4-二氢萘-1-酮、7-甲氧基-1-萘满酮、5-羟基-1-四氢萘酮、6-羟基-1-四氢萘酮与4-甲基-1-四氢萘酮均为分析纯,购自于上海麦克林生化科技有限公司,所有试剂使用前均未经进一步处理直接使用。去离子水为实验室自制二次水。数据分析采用:核磁共振仪,AVANCE NEO 400,布鲁克(北京)科技有限公司。
1.2 α-四氢萘酮肟及其同系物的合成
α-四氢萘酮肟及其同系物的合成如图1所示。
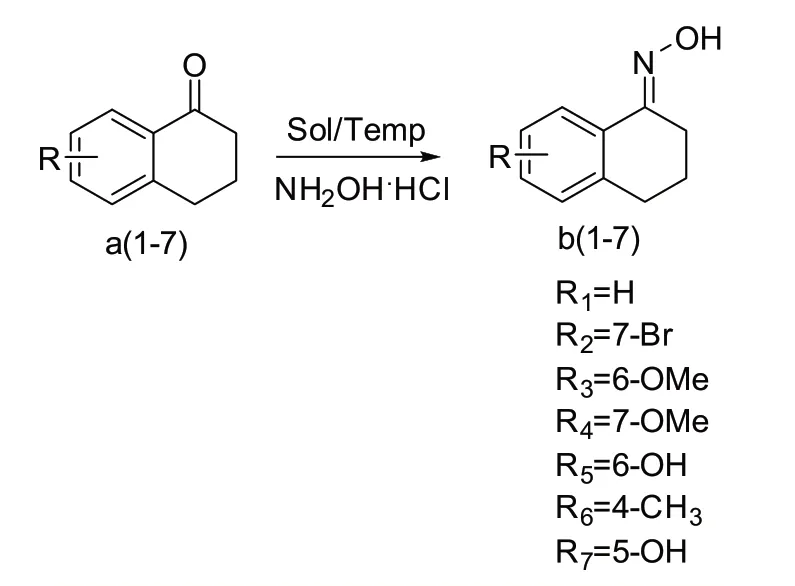
图1 酮肟结构的制备
3,4-二氢萘-1(2H)-一肟(b1):将6 mL无水乙醇和20 mL H2O配置成混合溶剂,再将α-四氢萘酮(0.4 g,2.7 mmol)加入到溶剂中,不断搅拌下依次加入盐酸羟胺(0.28 g,4.05 mmol)和乙酸钠(0.55 g,6.75 mmol),加热到95℃,搅拌反应约4 h,薄层色谱(展开剂:VDCM:VEA=20∶1)检测反应,原料α-四氢萘酮反应完全后,冷却至室温,再向反应溶液中加入4×10 mL乙酸乙酯萃取4次,将合并后的有机相减压蒸馏除尽有机溶剂,得到白色固体,然后将白色固体采用10 mL二氯甲烷和1 mL石油醚的混合溶剂重结晶,得到白色晶体b1(0.42 g,产率92%),1HNMR(400 MHz,CDCl3,ppm):δ9.05(s,1H),7.91(dd,J=7.8 Hz,1.1 Hz,1H),7.29(dd,J=7.6 Hz,1.3 Hz,1H),7.24(d,J=1.4 Hz,1H),7.19(dd,J=7.5,0.8 Hz,1H),2.87(t,J=6.7 Hz,2H),2.83~2.77(m,2H),1.97~1.87(m,2H)。
7-溴-3,4-二氢萘-1(2H)-一肟(b2)(采用与化合物b1类似的合成方法,下同):白色晶体,产率94%,
1HNMR(400 MHz,CDCl3,ppm):δ8.40(s,1H),8.07(d,J=2.1 Hz,1H),7.39(dd,J=8.2 Hz,2.1 Hz,1H),7.05(d,J=8.2 Hz,1H),2.83(t,J=6.7 Hz,2H),2.78~2.70(m,2H),1.94~1.83(m,2H)。
6-甲氧基-3,4-二氢萘-1(2H)-一肟(b3):浅黄色晶体,产率93%,1HNMR(400 MHz,CDCl3,ppm):δ9.07~8.61(m,1H),7.85(d,J=8.8 Hz,1H),6.80(dd,J=8.8 Hz,2.7 Hz,1H),6.69(d,J=2.6 Hz,1H),3.84(s,3H),2.84(t,J=6.6 Hz,2H),2.80~2.71(m,2H),1.94~1.85(m,2H)。
7-甲氧基-3,4-二氢萘-1(2H)-一肟(b4):黄色晶体,产率93%,1HNMR(400 MHz,CDCl3,ppm):δ8.86(s,1H),7.46(d,J=2.7 Hz,1H),7.09(d,J=8.4 Hz,1H),6.89(dd,J=8.4 Hz,2.7 Hz,1H),3.84(s,3H),2.83(t,J=6.7 Hz,2H),2.77~2.70(m,2H),1.89(dq,J=9.4 Hz,6.5 Hz,2H)。
6-羟基-3,4-二氢萘-1(2H)-一肟(b5):浅褐色晶体,产率87%,1HNMR(400 MHz,DMSO-d6,ppm):δ 10.65(s,1H),9.50(s,1H),7.68(d,J=8.6 Hz,1H),6.60(dd,J=8.6 Hz,2.5 Hz,1H),6.55(d,J=2.5 Hz,1H),2.61(dd,J=11.7 Hz,5.4 Hz,4H),1.77~1.66(m,2H)。
4-甲基-3,4-二氢萘-1(2H)-一肟(b6):白色晶体,产率93%,1HNMR(400 MHz,CDCl3,ppm):δ9.80(s,1H),8.01~7.89(m,1H),7.40~7.34(m,1H),7.32~7.28(m,1H),7.27(d,J=3.1 Hz,1H),2.97~2.91(m,2H),2.02(dddd,J=12.8,8.5,6.0,4.2 Hz,1H),1.75(td,J=12.9 Hz,6.8 Hz,1H),1.36(d,J=7.0 Hz,3H)。
5-羟基-3,4-二氢萘-1(2H)-一肟(b7):白色晶体,产率92%,1HNMR(400 MHz,DMSO-d6,ppm):δ10.95(s,1H),9.36(s,1H),7.34(dd,J=7.9 Hz,0.8 Hz,1H),6.97(t,J=7.9 Hz,1H),6.76(dd,J=7.9 Hz,1.0 Hz,1H),2.65~2.61(m,2H),2.61~2.57(m,2H),1.77~1.65(m,2H)。
1.3 α-四氢萘酮肟酸酯及其同系物的合成
α-四氢萘酮肟酸酯及其同系物的合成如图2所示。
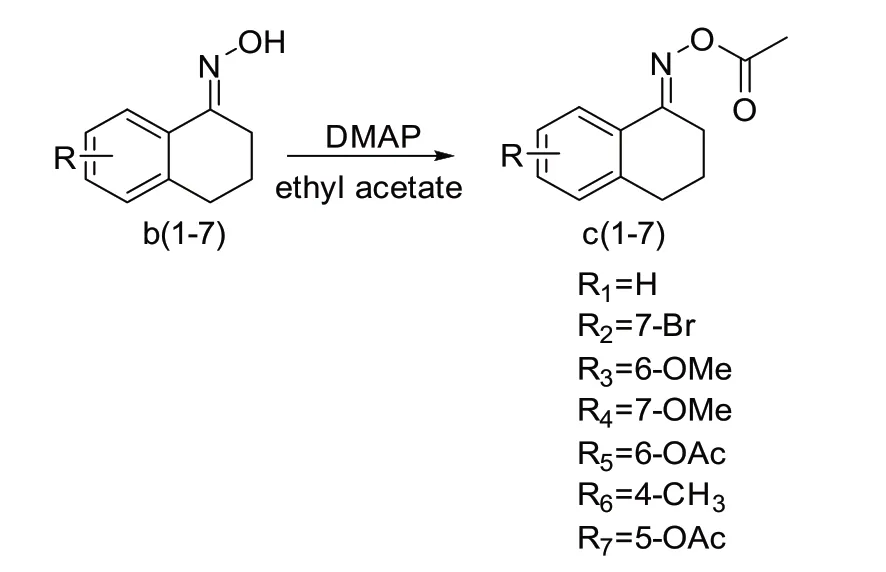
图2 酮肟制备酮肟酸酯
3,4-二氢萘-1(2H)-一肟酸酯(c1):将3,4-二氢萘-1(2H)-一肟(0.4 g,2.48 mmol)加入到20 mL乙酸乙酯中,不断搅拌溶解,溶解完全后,加入乙酸酐(0.35 mL,2.72 mmol),持续搅拌下,缓慢加入4-二甲氨基吡啶(0.06 g,0.496 mmol),反应约0.5 h,薄层色谱(展开剂:VDCM:VEA=20∶1)检测反应,待反应完全后,向反应液中加入10 mL水淬灭反应,分液,水相采用10 mL乙酸乙酯再萃取一次,合并后的有机相,减压蒸馏除尽溶剂,得到白色固体粗产品,然后使用约5 mL二氯甲烷重结晶,得到白色针形晶体c1,产率81%,m.p.:118℃。1HNMR(400 MHz,CDCl3,ppm):δ8.16(dd,J=7.9,1.0 Hz,1H),7.36(td,J=7.5,1.4 Hz,1H),7.25(ddd,J=7.9,1.3,0.7 Hz,1H),7.19(dd,J=7.6,0.7 Hz,1H),2.93~2.86(m,2H),2.85~2.78(m,2H),2.29(s,3H),1.96~1.87(m,2H)。13CNMR(101 MHz,CDCl3,ppm):δ169.18(s),161.28(s),140.92(s),130.72(s),128.98(s),128.70(s),126.57(s),125.60(s),29.54(s),25.58(s),21.34(s),19.86(s)。
7-溴-3,4-二氢萘-1(2H)-一肟酸酯(c2)(采用与化合物c1类似的合成方法,下同):白色晶体,产率79%,m.p.:127℃。1HNMR(400 MHz,CDCl3,ppm):δ8.29(d,J=Hz,1H),7.46(dd,J=8.2,2.1 Hz,1H),7.07(d,J=8.2 Hz,1H),2.90~2.82(m,2H),2.79~2.72(m,2H),2.29(s,3H),1.90(dq,J=9.1,6.5 Hz,2H)。13CNMR(101 MHz,CDCl3,ppm):δ168.89(s),160.21(s),139.69(s),133.58(s),130.84(s),130.33(s),128.21(s),120.40(s),29.07(s),25.31(s),21.05(s),19.81(s)。
6-甲氧基-3,4-二氢萘-1(2H)-一肟酸酯(c3):淡黄色晶体,产率80%,m.p.:84℃。1HNMR(400 MHz,CDCl3,ppm):δ8.11(d,J= 8.8 Hz,1H),6.80(dd,J=8.8,2.7 Hz,1H),6.69(d,J=2.6 Hz,1H),3.84(s,3H),2.85(d,J=6.6 Hz,2H),2.78(s,2H),2.27(s,3H),1.95~1.85(m,2H)。13CNMR(101 MHz,CDCl3,ppm):δ169.24(s),161.53(s),161.11(s),142.89(s),127.40(s),121.57(s),113.02(s),112.97(s),55.28(s),29.87(s),25.47(s),21.40(s),19.87(s)。
7-甲氧基-3,4-二氢萘-1(2H)-一肟酸酯(c4):黄色晶 体,产 率78%,m.p.:67℃。1HNMR(400 MHz,CDCl3,ppm):δ7.64(d,J=2.7 Hz,1H),7.09(d,J=8.4 Hz,1H),6.94(dd,J=8.4,2.8 Hz,1H),3.84(s,3H),2.89~2.81(m,2H),2.76~2.69(m,2H),2.27(s,3H),1.92~1.82(m,2H)。13CNMR(101 MHz,CDCl3,ppm):δ169.12(s),161.49(s),158.12(s),133.57(s),129.72(d,J=7.4 Hz),118.70(s),108.48(s),55.52(s),28.75(s),25.48(s),21.57(s),19.86(s)。
6-乙酸酯基-3,4-二氢萘-1(2H)-一肟酸酯(c5):褐色晶体,产率77%,m.p.:131℃。1HNMR(400 MHz,CDCl3,ppm):δ8.17(d,J=8.5 Hz,1H),6.95(dt,J=4.1,2.1 Hz,2H),2.89~2.81(m,2H),2.81~2.73(m,2H),2.29(s,3H),2.25(s,3H),1.93~1.82(m,2H)。13CNMR(101 MHz,CDCl3,ppm):δ169.09(d,J=4.7 Hz),160.54(s),152.40(s),142.52(s),127.18(s),126.69(s),121.46(s),120.05(s),29.56(s),25.40(s),21.13(d,J=3.0 Hz),19.83(s)。
4-甲基-3,4-二氢萘-1(2H)-一肟酸酯(c6):白色晶体,产率82%,m.p.:78℃。1H NMR(400 MHz,CDCl3,ppm):δ8.18~8.13(m,1H),7.40(td,J=7.8,1.4 Hz,1H),7.28(s,1H),7.27(d,J=3.6 Hz,1H),3.00~2.92(m,2H),2.29(s,3H),2.06~1.95(m,1H),1.72(s,1H),1.33(d,J=7.0 Hz,3H)。13CNMR(101 MHz,CDCl3,ppm):δ169.18(s),161.33(s),145.61(s),130.95(s),128.29(s),127.11(s),126.47(s),125.67(s),32.70(s),28.37(s),22.79(s),20.39(s),19.87(s)。
5-乙酸酯基-3,4-二氢萘-1(2H)-一肟酸酯(c7):浅褐色晶体,产率80%,m.p.:124℃。1HNMR(400 MHz,CDCl3,ppm):δ8.09(dd,J=8.0 Hz,1.0 Hz,1H),7.26(t,J=8.3 Hz,1H),7.12(dd,J=8.0,1.2 Hz,1H),2.89~2.83(m,2H),2.67~2.60(m,2H),2.34(s,3H),2.27(s,3H),1.92~1.83(m,2H)。13CNMR(101 MHz,CDCl3,ppm):δ169.00(d,J=6.1 Hz),160.64(s),148.37(s),133.16(s),130.70(s),126.89(s),124.11(s),123.43(s),25.02(s),22.88(s),20.74(s),20.51(s),19.83(s)。
2 结果与讨论
2.1 α-四氢萘酮肟及其同系物合成分析
首先从溶剂、反应温度与碱催化剂3个影响因素来探索α-四氢萘酮及其衍生物合成α-四氢萘酮肟的最优条件,其中反应时间为4 h,不同条件下产率见表1。
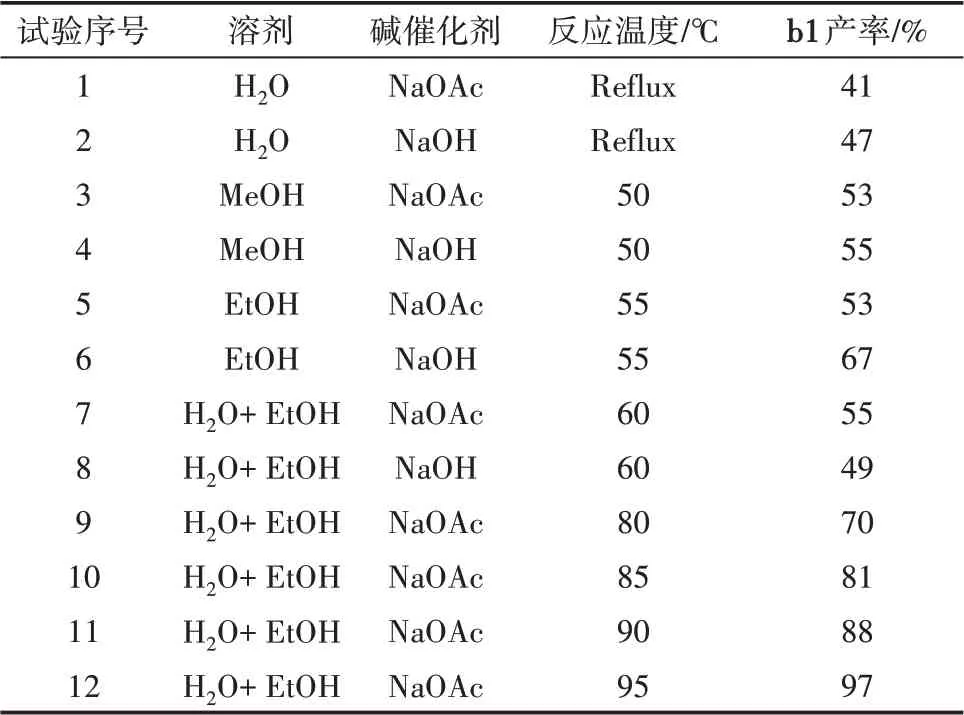
表1 合成α-四氢萘酮肟及其衍生物的条件探索
α-四氢萘酮肟及其同系物的合成实验中:当α-四氢萘酮作为反应底物,以水作为溶剂,回流条件下反应4 h,考察NaOH与NaOAc两种碱存在下α-四氢萘酮与盐酸羟胺的反应,两者反应收率差别不大,NaOH的效果要略优于NaOAc(表1,试验1—2)。在相同温度和时间的条件下,乙醇作为溶剂时反应收率比甲醇作为溶剂时的收率要高,可能是因为乙醇的溶解效果更好,而在单一溶剂的情况下NaOH的催化效果要略优于NaOAc,产物的收率要高(表1,试验3—6)。由于盐酸羟胺易溶于醇,而乙酸钠又易溶于水,于是进行了在混合溶剂中的反应。以水和乙醇为溶剂条件下反应4 h,可以看出NaOAc的效果要优于NaOH,这可能是因为混合溶剂的条件下氢氧化钠对盐酸羟胺的影响较大,消耗了盐酸羟胺,导致产率不如乙酸钠(表1,试验7—8)。进一步,考察了乙酸钠为碱的条件下温度对该反应的影响(表1,试验9—12)。从表1可以看出,相同溶剂、时间和碱的条件下,温度升高,产率增大,这可能是因为温度升高,酮与盐酸羟胺的反应更加迅速,原料分解更少。最后得出最佳反应条件:以水和乙醇作为反应溶剂,乙酸钠作为碱催化剂,在95℃下反应4 h。
2.2 α-四氢萘酮肟酸酯及其同系物合成分析
制备得到酮肟结构中间体后,再对肟制备酮肟酸酯的反应条件进行了探索,见表2。其中反应温度均为室温。
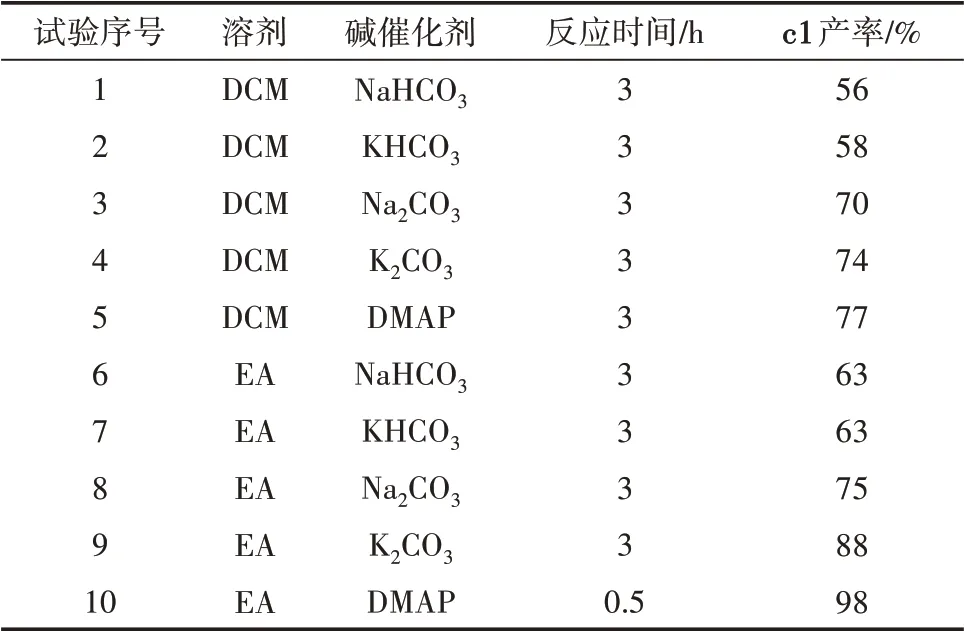
表2 α-四氢酮肟酸酯的合成条件探索
α-四氢酮肟酸酯及其同系物的合成实验中,当α-四氢萘酮肟作为反应底物,乙酸酐作为酯化试剂,以二氯甲烷作为溶剂,室温条件下反应3 h。首先考察了NaHCO3、KHCO3、K2CO3、Na2CO3和DMAP 5种碱对反应的影响(表2,试验1—5),结果表明DMAP的效果最佳,K2CO3、Na2CO3的效果次之,NaHCO3、KHCO3的效果最差,这可能是因为碳酸氢钠和碳酸氢钾的碱性太弱,因此碱性强有助于反应的进行,而DMAP作为有机碱更易溶于有机溶剂,其他四种碱为无机碱,溶解性差,因而DMAP催化效果最佳。进一步地,考察了乙酸乙酯作为溶剂时合成α-四氢酮肟酸酯及其同系物的反应效果(表2,试验6—10),可以看出乙酸乙酯作为反应溶剂时5种碱的效果普遍优于二氯甲烷时的效果,可能是乙酸乙酯与二氯甲烷的极性不同以及对于5种碱的溶解性差异。因此综合考虑上述探索结果,选择乙酸乙酯作为反应溶剂,DMAP为碱,室温反应0.5 h,作为合成酮肟酸酯及其衍生物的最佳工艺条件。
3 结束语
肟酸酯及其衍生物是一类重要的合成中间体,研究其合成工艺具有理论及实际应用价值。通过研究得到了一种合成酮肟酸酯的新路线:以α-四氢萘酮及其同系物作为原料,与盐酸羟胺进行反应得到α-四氢萘酮肟;然后以α-四氢萘酮肟作为反应底物,与乙酸酐反应制备酮肟酸酯,两步反应总收率在70%以上。该方法具有原料易得、反应条件温和及反应收率高等优点。主要从溶剂、反应时间、温度和碱催化剂的种类等方面考察了制备肟和肟酸酯的反应条件,最后得到最佳反应条件为:在H2O和EtOH为混合溶剂、乙酸钠为碱的条件下与盐酸羟胺在95℃下反应4 h制备中间体酮肟结构,产率能达到90%以上;然后以该中间体为底物,在室温条件下,采用DMAP为催化剂,与乙酸酐反应0.5 h制备得到酮肟酸酯,最终产率能达到75%以上。以上研究得到的合成方法具有反应条件温和、不产生重金属盐废渣、产物收率较高等特点。
猜你喜欢
能源与环境(2022年5期)2023-01-10
城市道桥与防洪(2022年3期)2022-05-08
昆明医科大学学报(2021年8期)2021-08-13
安全与环境工程(2021年2期)2021-04-02
现代牧业(2020年4期)2020-12-31
煤炭加工与综合利用(2020年6期)2020-07-17
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中成药(2017年3期)2017-05-17
军事文摘·科学少年(2016年10期)2016-12-08
