
Structural modeling of Nav1.5 pore domain in closed state
2021-12-30 13:26XiaofengJiYanzhaoHuangJunSheng
Biophysics Reports 2021年4期
Xiaofeng Ji ,Yanzhao Huang,Jun Sheng
1 Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, Shandong, China
2 School of Physics, Huazhong University of Science and Technology, Wuhan 430074, China
Abstract The voltage-dependent cardiac sodium channel plays a key role in cardiac excitability and conduction and it is the drug target of medically important.However, its atomic- resolution structure is still lack.Here, we report a modeled structure of Nav1.5 pore domain in closed state.The structure was constructed by Rosetta-membrane homology modeling method based on the template of eukaryotic Nav channel NavPaS and selected by energy and direct coupling analysis (DCA).Moreover, this structure was optimized through molecular dynamical simulation in the lipid membrane bilayer.Finally, to validate the constructed model, the binding energy and binding sites of closed-state local anesthetics (LAs)in the modeled structure were computed by the MM-GBSA method and the results are in agreement with experiments.The modeled structure of Nav1.5 pore domain in closed state may be useful to explore molecular mechanism of a state-dependent drug binding and helpful for new drug development.
Keywords Nav1.5 ion channel, Closed state, Homology modeling, Direct coupling analysis, Local anesthetics, Drug binding
INTRODUCTION
In the early stage, due to the lacking of experimental structures of Navchannel, all modelled structures were modeled by using potassium channels as templates(Lipkind and Fozzard 2000; O’Reillyet al.2012).The structures of bacterial Navchannels, including NavAb(Payandehet al.2011, 2012), NavMs (Bagneriset al.2014; McCuskeret al.2012), and NavRh (Zhanget al.2012), were successively resolved and they revealed that the Navchannels were formed by four domains (DI to DIV) and each domain consisted of six transmembrane segments.In each domain, voltage sensor domain (VSD) was composed by the first four transmembrane segments (S1 to S4) and the ion conducting pore domain (PD) was formed by the other two segments (S5 and S6), with the pore loop (P-loop)between them.Based on these structures, Nav1.5 models were also reported (Ahmedet al.2017; Moreauet al.2015; Poulinet al.2014; Wanget al.2015; Xiaet al.2013).Recently, we also constructed an open-state structure of Nav1.5 by using the bacterial NavMs as template (Jiet al.2018).As we all know, the bacterial Navchannel shares similar overall architecture with eukaryotic one but differs in that the former is homotetramer while the latter is non-homo-tetramer.Recently, the first structure of eukaryotic Navchannel(NavPaS) at near-atomic resolution has been resolved(Shenet al.2017) and this makes it possible to build structures of eukaryotic Nav channels with higher precision.
Homology modeling methods have been widely used to construct 3D models of proteins from their amino acid sequences (Korkoshet al.2014; Lafitaet al.2018).These methods were based on the similarity of the sequence of the target protein to other proteins whose 3D structures were known.The Rosetta membrane homology modeling method (Sidharthaet al.2010;Subbotinaet al.2010) were used for constructing 3D models of membrane proteins.It searched for fragments with similar sequences in 3D structure databases and could predict membrane protein structure with reasonable accuracy (Subbotinaet al.2010).
In the structural modeling, information of residue-residue contacts will facilitate to find out the best conformation from the modeled decoys by combining with energy score, but experimental information of residue-residue contacts is usually unknown in advance.Therefore, the prediction of the contacts from sequences will be very helpful.The direct coupling analysis (DCA) of co-evolutionary residues has been shown to be an efficient method to do this(Jarzynski 2012; Luntet al.2010; Morcoset al.2014;Morcoset al.2011; Weigtet al.2009).DCA gives a score for each pair of residues in an amino-acid sequence or between two sequences to evaluate if the two residues can form contact.
In this work, we will model closed-state structures of Nav1.5 pore domain (PD) based on the template of eukaryotic Navchannel NavPaS (Shenet al.2017) and apply RosettaMP energy and DCA to select the rational structures of monomers and PD.We will validate the modeled structures by docking closed-state local anesthetics (LAs) to it and compare their binding energies and binding sites with previous experimental results.
RESULTS AND DISCUSSION
EC-score calculation for the sodium channel membrane proteins with known structures
We first investigate whether the co-evolutionary residues of intra- and inter-domains of Navchannels do exist in the tertiary structure by assessing blinded predictions of residue co-evolution against the determined structures NavAb (PDB ID: 3rvy), NavPaS(PDB ID: 5x0m) and NavRh (PDB ID: 4dxw).The results of the multiple sequence alignments were listed in supplementary Table S1.For each pair of residues within or across the domains, EVcouplings calculates an EC score for it, which describes the degree of coevolution of the two residues.Because the four domains of NavAb and NavRh have the same sequence,but the four domains of NavPaS are different, we only calculate the EC score and residue-residue distances of one domain for NavAb and NavRh, and the EC score and residue-residue distances for NavPaS.Figure 1 shows the correlation between EC scores and residue-residue distances of intra-domains.Here we define that a pair of residues with an 8 Å minimum atom distance between them are in contact.From Fig.1, we can find that the distance of two residues is usually less than 8 Å when the EC score is large than 0.2.Therefore, if the EC score of two residues is more than 0.2, the two residues are considered to form contact in the 3D structure.
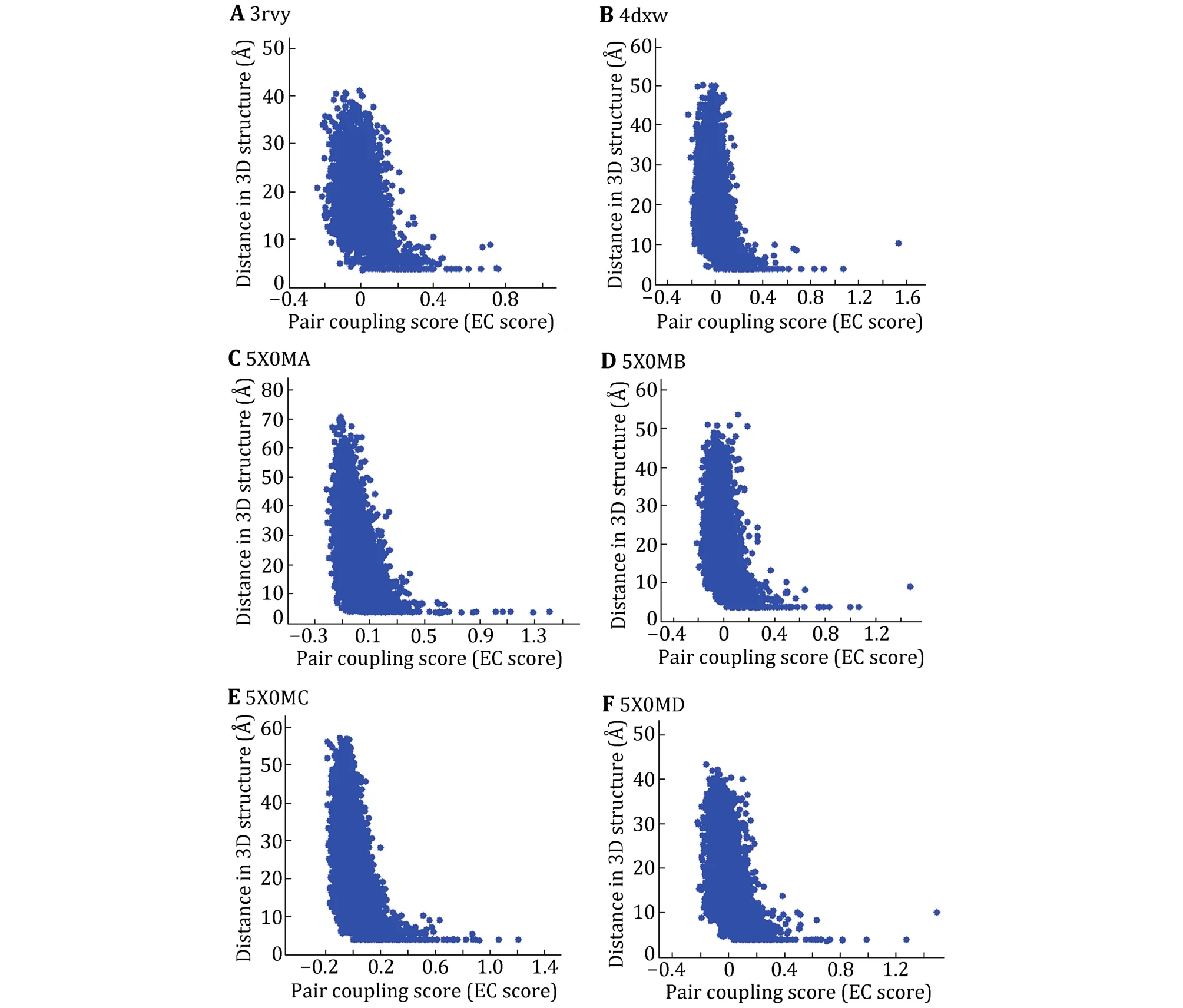
Fig.1 Maps of EC score and residue-residue distances for the three Nav channels with known tertiary structure.3rvy (A), 4dxw (B) and the four domains of 5x0m (C-F).X axis is EC score and Y axis is residue-residue distance in the 3D structure
Based on this threshold (0.2), the intra- and intercontact pairs of the domains are predicted and the number of predicted contact pairs is labeled as CA_DCA.Number of contact pairs in 3D structure is labeled as CA_Str.To access the accuracy rate of the predicted contacts, we investigated the coincidence of the predicted contact pairs and those in 3D structure.Table 1 shows that among 52 predicted contact pairs in single domain, there are 32 contact pairs that do have contacts in the 3D structure of NavAb and the rate of correctly predicted is nearly 62%.In particular, among the predicted contact pairs with top ten EC scores,there are eight pairs in the 3D structure.Similar to NavAb, the accuracies for NavRh and NavPaS are more than 50% and for 5x0mD it is even more than 69%.Because among the three Navchannels with known structures, only the domains of NavPaS have distinct conformations, it was chosen to be the evaluation protein to access whether the EC scores can be used to predict contact residues between protein domains.The inter-EC pairs for NavPaS are listed in Table 2.Compared to intra-contacts, the accuracy of inter-contact prediction is very lower.For the adjacent domains (AB, BC, CD and AD), the precision is about 10%.These results demonstrate that the EC scores can be used to determine contacts in one of the domains of Navchannels but are not good enough for inter-contacts between the domains.However, the latter can also be considered as a reference since we can see from Table 2 that the precisions of the predicted contacts between the adjacent domains are much higher than those of nonadjacent domains.By the way, these results also indicate that the formation of the tetramers of Navchannels may not be determined mainly by the specific interactions of the co-evolved residues between the domains but by other interactions.
Two or three days later the master said to him: The Porcelain Maiden is here: but, though she is as lovely as the dawn, she is so wicked that she scratches everyone that approaches her
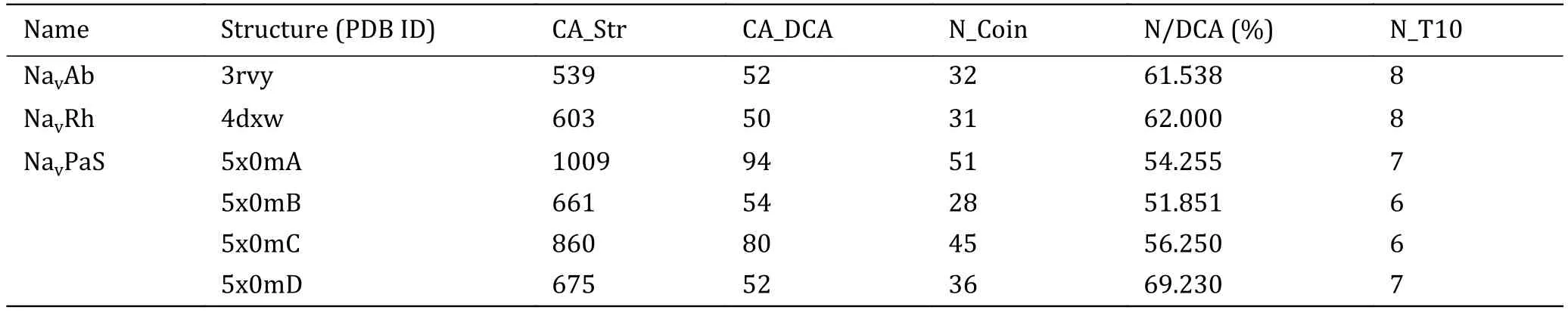
Table 1 Intra-EC pairs for the three Nav channels with known tertiary structure
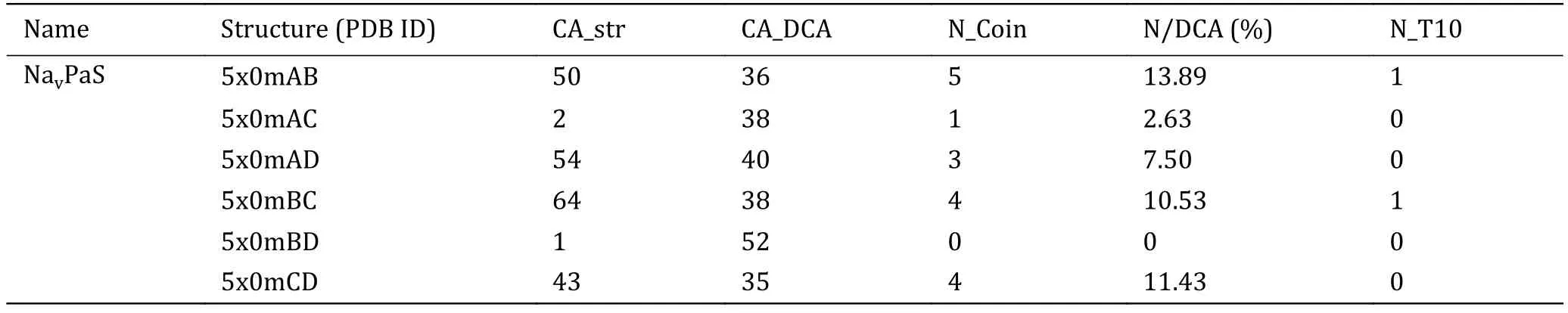
Table 2 Inter-EC pairs for NavPaS
Structural modeling and selection of Nav1.5 domains
Using sequences of the four domains of Nav1.5 as the input, we searched the database of Protein Data Bank(PDB, www.rcsb.org.com) using BlastP.From the search results, we found that compared to other four bacterial Navchannels (NavAb, NavRh and NavMs,NavPaS), all of the four target sequences had the highest sequence identities (48%) with the corresponding domains of NavPaS.Meanwhile, NavPaS was the sole eukaryotic sodium ion channel with known tertiary structure by far and it had a closed pore.Hence, the structure of NavPaS was chosen as the template structure and the ROSETTA membrane-homology modeling method was used to model the PD structure of Nav1.5 channel in closed state.The four target sequences were aligned to corresponding sequences of NavPaS by HHpred program with the parameters as system default (E-value cutoff 1 × 10-3, minimum coverage of multiple sequence alignment hits 20%,realignment threshold 0.3).The alignment results(supplementary Fig.S1) show that the insertion and deletion are mainly in P-segments but not any in S5 and S6.For each domain, 10,000 models were generated.Then, we calculated their energies by using RosetaMP and the distances between any two residues.We also calculated the EC scores of each residue pairs by using EVcouplings.For the four domains, 1939, 3923, 3390 and 3597 homologous sequences were used to generate the multiple sequence alignment to calculate EC scores, respectively.Table 3 shows the intra-EC pairs for the four domains of Nav1.5.From Table 3, we can find that for domain 1, structure domain1_5x0mS_0177 and domain1_5x0mS_0467 are the top two structures with the highest energy score and precision of contact prediction,i.e.these two structures not only have the lowest energy, but also have the highest consistency of the residue-residue contacts with the DCA prediction.Hence these two structures were selected as the modeled structures of domain 1.Similar to this procedure, the modeled structures for other three domains were selected (the bold in Table 3).Meanwhile, we can find out that these selected structures have the lowest energy as well as higher precision of contact prediction.The Ramachandran plots of the selected models also show that the dihedral angles of all residues are located in the allowed regions (supplementary Fig.S2).The helices S5 and S6 of the selected models are nearly the same and the differences occur in the region of P-segments(supplementary Fig.S3).
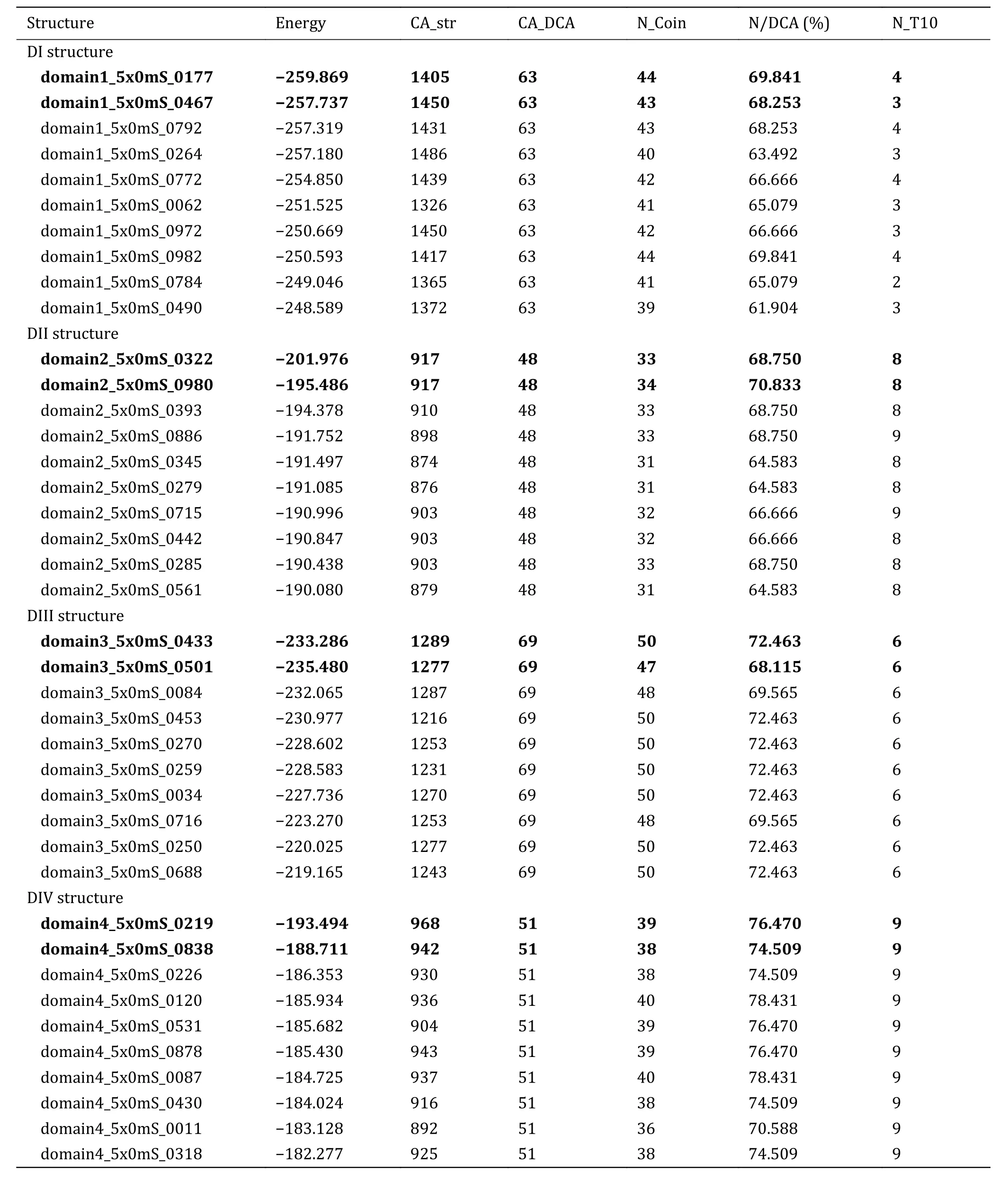
Table 3 Candidates for structures of the four domains of Nav1.5
Finally, 16 PD models were generated by using the selected models of the four domains and scored by RosettaMP.From Table 4, we can see that the RosettaMP energy of closed10 is the lowest.It is noted that the N/DCA value of closed10 is also the highest although its absolute value is low.Therefore, the closed10 is selected for subsequent research (the bold in Table 4).
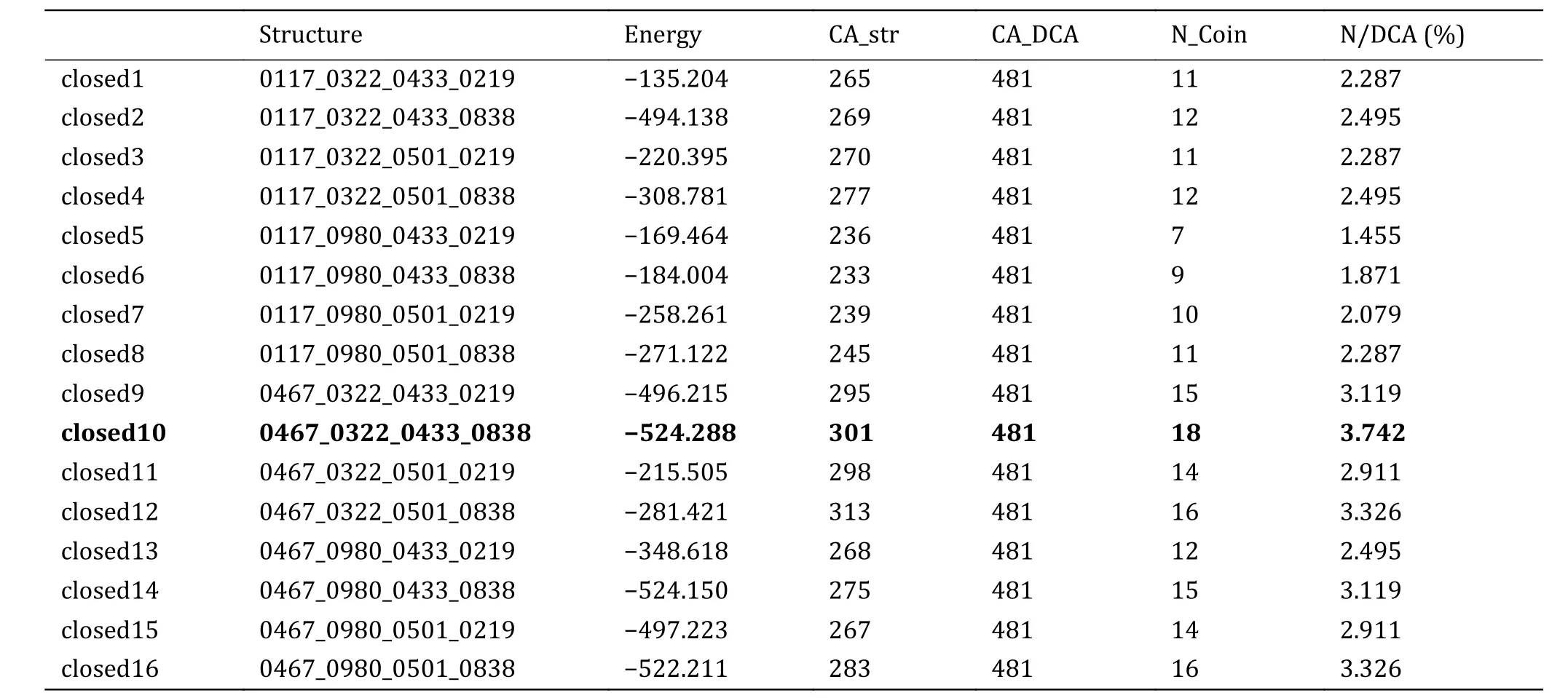
Table 4 Energy and inter-domain EC pairs of 16 PD models
To destroy atom clashes and tensions, the close10 is further optimized by a 450-ns MD simulation and finally reaches an equilibrated structure with a fluctuated outer-membrane P-loops (Fig.2A and supplementary Fig.S4).The Ramachandran plot also shows that the dihedral angles of almost all residues in the equilibrated structure are located in the allowed regions (Fig.2B).Figures 2C and 2D show the equilibrated structure from different views.Figures 2E and 2F show the channel and its radius versus distance along the channel direction for the equilibrated structure, respectively.They show that the diameter of the gate region is about 3.48 Å, which is much smaller than that of sodium ions (roughly 7.8 Å)and so sodium ions cannot pass through the channel in this case.This means that the constructed model is in closed state.Furthermore, Figures 3A and 3B show the selective filter (SF) of the close10 formed by the side groups of residues D121/E225/K356/A463 and the carbonyl oxygen atoms of their two preceding residues.
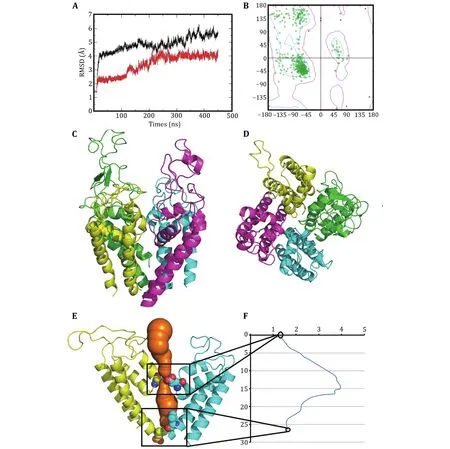
Fig.2 Optimized structure of the closed10.A The changes of the RMSDs of Cα atoms of the closed10 with (black line) and without (red line) outer membrane P-loops during 450-ns MD simulation.B Ramachandran plot of the optimized structure.The side (C) and top (D) views of the optimized structure, generated by pymol (Lilkova et al.2015) and colored by chain (green, cyan, purple and yellow for D1-D4).The channel (in orange) (E) and its radius versus distance along the channel direction (F)
The outer negative ring that guards the entrance to the SF vestibule is formed by E124/E228/M359/D466.Compared to NavPaS, the differences are mainly in the orientation of residue K356 and D121 (Fig.3C and 3D).Aligned the structure of the closed10 without outer membrane to its open state that we had constructed before by using TM-align (Zhang and Skolnick 2005), the result shows that the RMSD is 3.77 and the TM-score is 0.41606.Furthermore, in order to check the stability of the modelled structure, we calculated the diameter of the gate region and the all atoms RMSF during the simulation.For diameter calculation, the larger distance of D121-K356 and E225-A463 is defined as the diameter of the gate region of the corresponding frame.The results show that during the simulation, the diameter of the gate region is in the range of 4.19 to 1.21(supplementary Fig.S5A), which is smaller than that of sodium ions,i.e.the door is closed during the simulations.RMSF values were then calculated to analyze the fluctuations of all residues (supplementary Fig.S5B).In supplementary Fig.S5B, the higher values are found in regions of loops and the connection areas of the domains.This means that the positions of residues in regions of S5, S6 and P-loops in the four domains are almost unchanged.
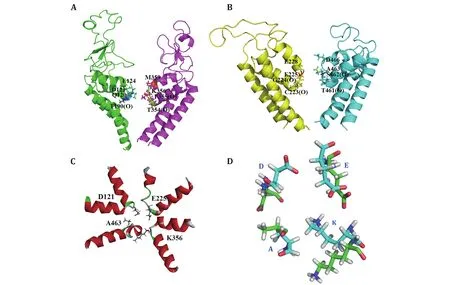
Fig.3 The selectivity filter (SF) of the model closed10.A, B The SF residues D121/E225/K356/A463 with their two preceding residues and the residues that constitute the negative ring above the SF vestibule.All these residues are represented in stick.C SF top view is shown.D SF structural variations of closed10 (green) and NavPaS (cyan)
Structure validation
Previous studies show that the inner vestibule of Nav1.5 PD is the binding pocket of open-state and closed-state LA drugs (Lipkind and M 2005) and the residues L214(L1462 in Nav1.5α) in S6 of D3, and F512 (F1760 in Nav1.5α) and Y519 (Y1767 in Nav1.5α) in S6 of D4 are parts of their binding sites.The closed-state LAs include Pilsicainide, Bisphenol A and Mexiletine, and the binding strength of them has been measured by experiments.Therefore, this information can be used to validate the closed10 model by finding the binding sites and binding energies of the closed-state LAs with the closed10.
We docked the three closed-state LAs to the closed10.Three independent dockings were made and the conformations of LAs were clustered with the cutoff of 2 Å (Table 5).Mapping all of these conformations to the closed10 (Fig.4), it can be found that all of them are located within the cavity enclosed by S6 segments.This result is consistent with previous reports.For Pilsicainide, Bisphenol A and Mexiletine, the clusters C1, C4 and C2 have the lowest average binding energy respectively, and the conformations with the lowest binding energy are also in these clusters.These three lowest energy conformations are chosen to select their complexes with the closed10, respectively.
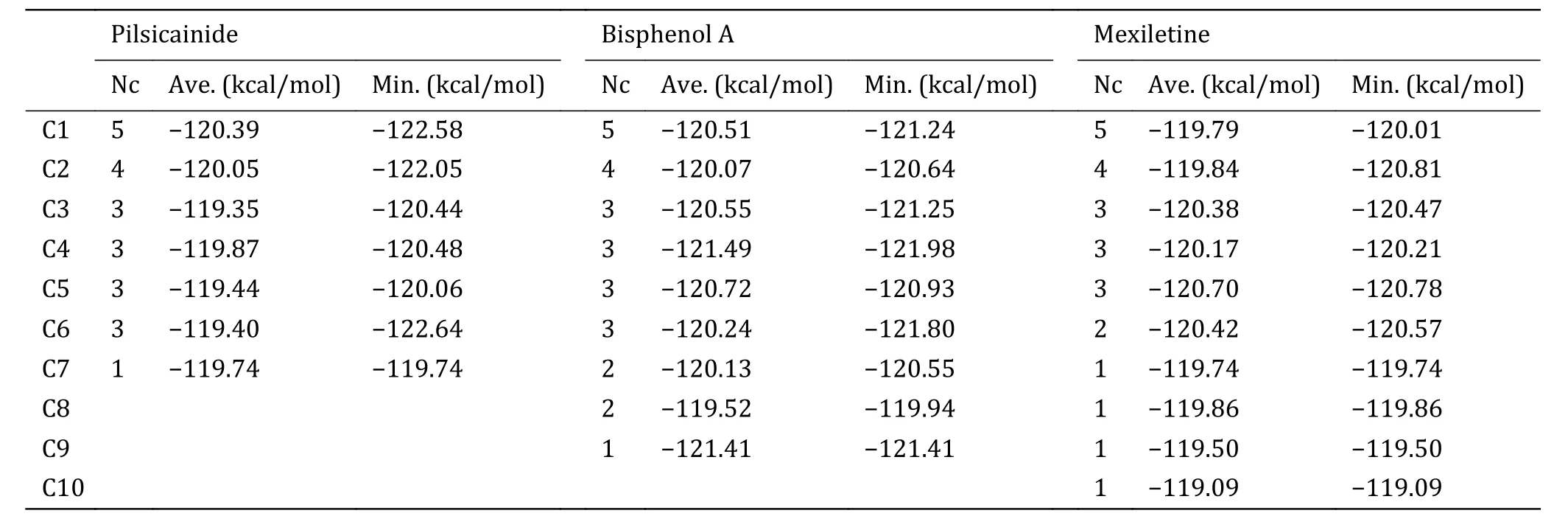
Table 5 Cluster results of conformations of three independent dockings for Pilsicainide, Bisphenol A and Mexiletine
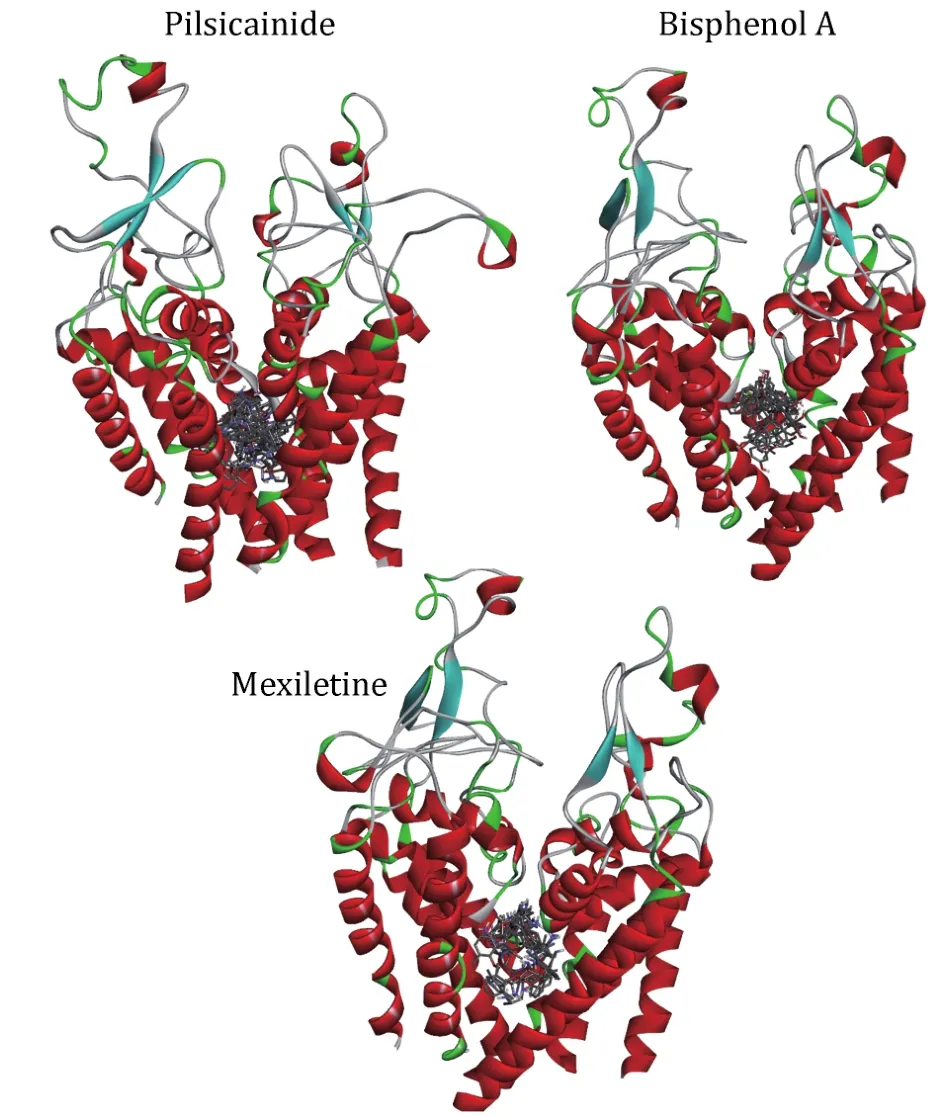
Fig.4 The relative position of docking conformations of LAs with the closed10.The plot is generated by DS Visualizer 4.2, protein structures are shown in cartoon and drugs are shown in sticks
In order to detect the ability of our model to discriminate strong from weak closed-state LAs, we calculated the binding energies of LA-closed10 complexes.For each complex, 300-ns MD simulation was done and the last 30 ns (3 × 103frames) was used to calculate the binding energy (Fig.5).In order to clarify the dynamic stability of these structures, RMSD values were obtained using the initial structures as templates for ligands (supplementary Fig.S6).The L_RMSD plots show that the RMSD values of the atoms (in nofit) with respect to the initial structures increased for 150 ns.After that, they remained fluctuated around 1.7-2 Å until the end of the simulation.Thus, the trajectories of the MD simulations of these structures were reliable.
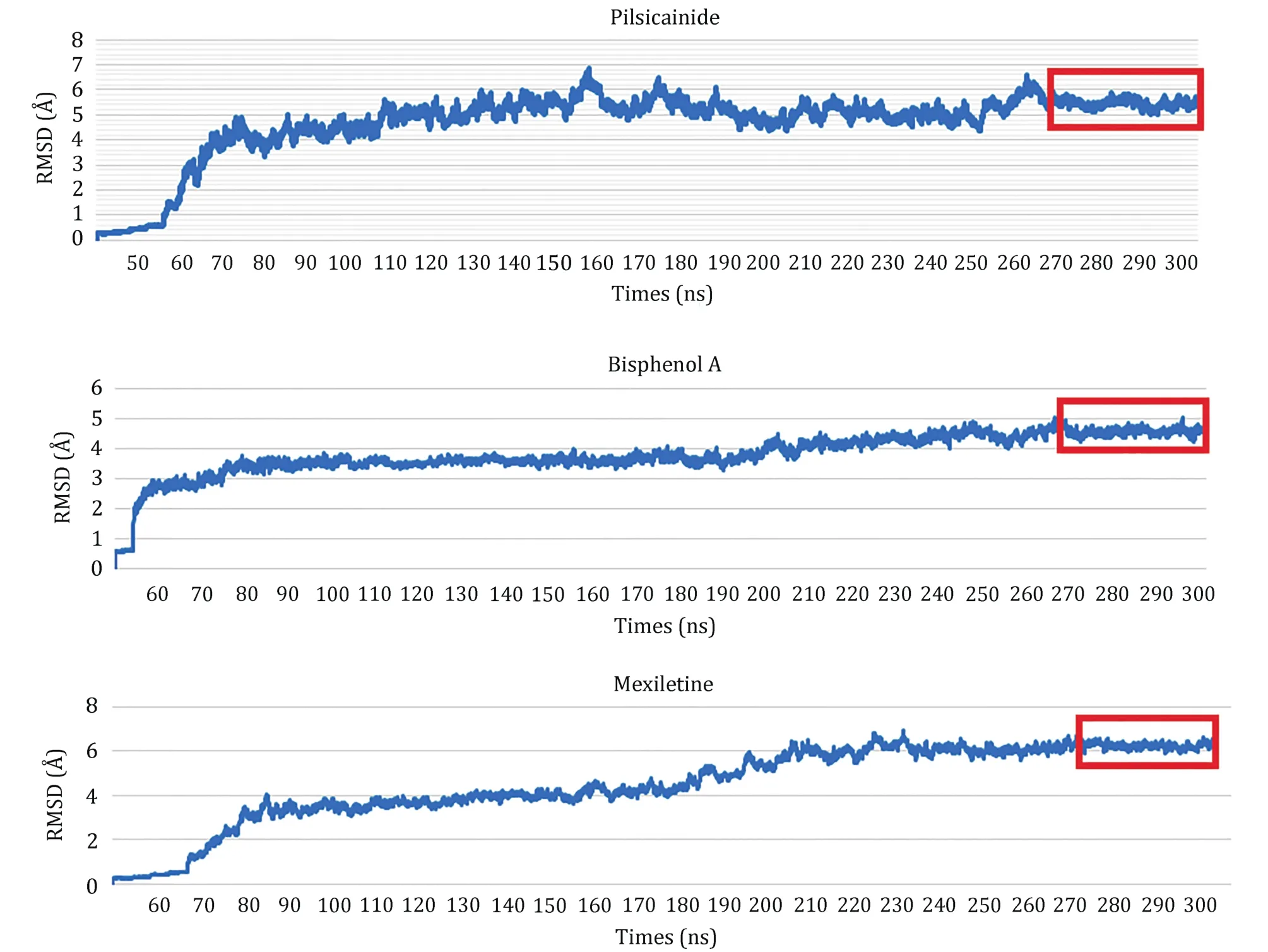
Fig.5 RMSDs for the complexes of Nav1.5_Las.The trajectories in the red rectangles are used to calculate binding energies
The binding energies calculated by MM-GBSA are summarized in Table 6.It can be found that polar solvation plays positive role while no-polar solvation plays negative role for interaction of LAs and Nav1.5,i.e.van der Waals, electrostatic and accessible surface area facilitate the binding of LAs with Nav1.5 while the polar solvation destabilizes their binding.Totally, the closed10 binds more tightly with Mexiletine than the other two Las, and binds most loosely with Pilsicainide.This result is consistent with the previous reports aboutKdvalues of these three LAs (Liet al.1999; Reillyet al.2012; Plesset al.2011).
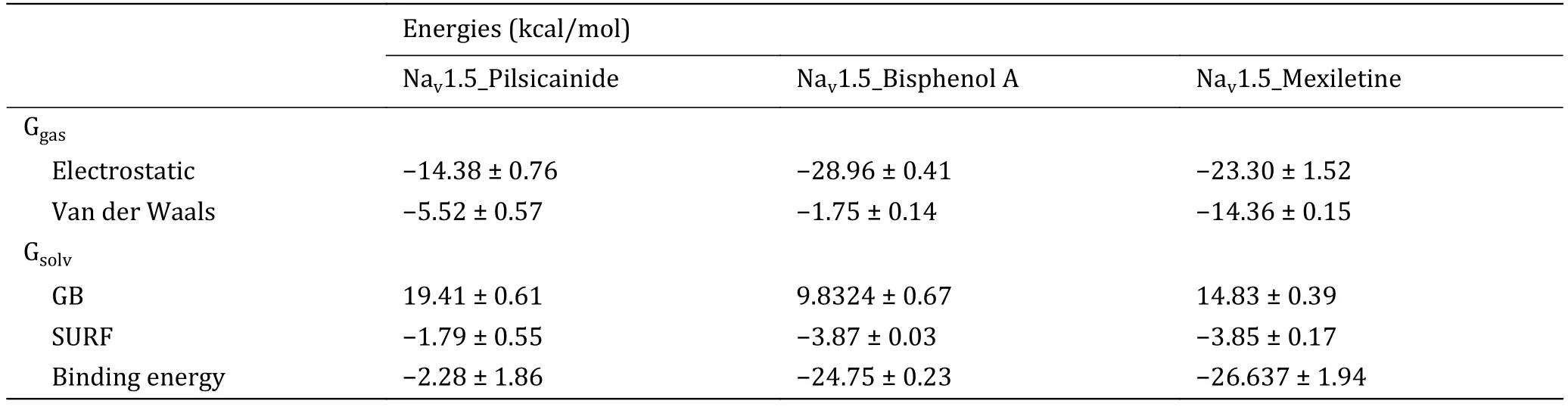
Table 6 Binding energies of LA_closed10 complexes
To further investigate the binding sites of LAs, the binding energies are decomposed to each residue(Fig.6).For Mexletine, the interaction energy of D121(-3.0035 kcal/mol), T416 (-2.001 kcal/mol), A463(-2.202 kcal/mol) and F512 (-3.29 kcal/mol) are less than -2.0 kcal/mol.Of these four residues, the contribution of F512 (F1760 in Nav1.5α) is the most significant, which is consistent with the previous report that this residue is the key residue of LA binding with Nav1.5.According to the analysis of the interaction type(Fig.7C), the interaction between F512 and Mexletine is mainly through the pi-alkyl interaction.
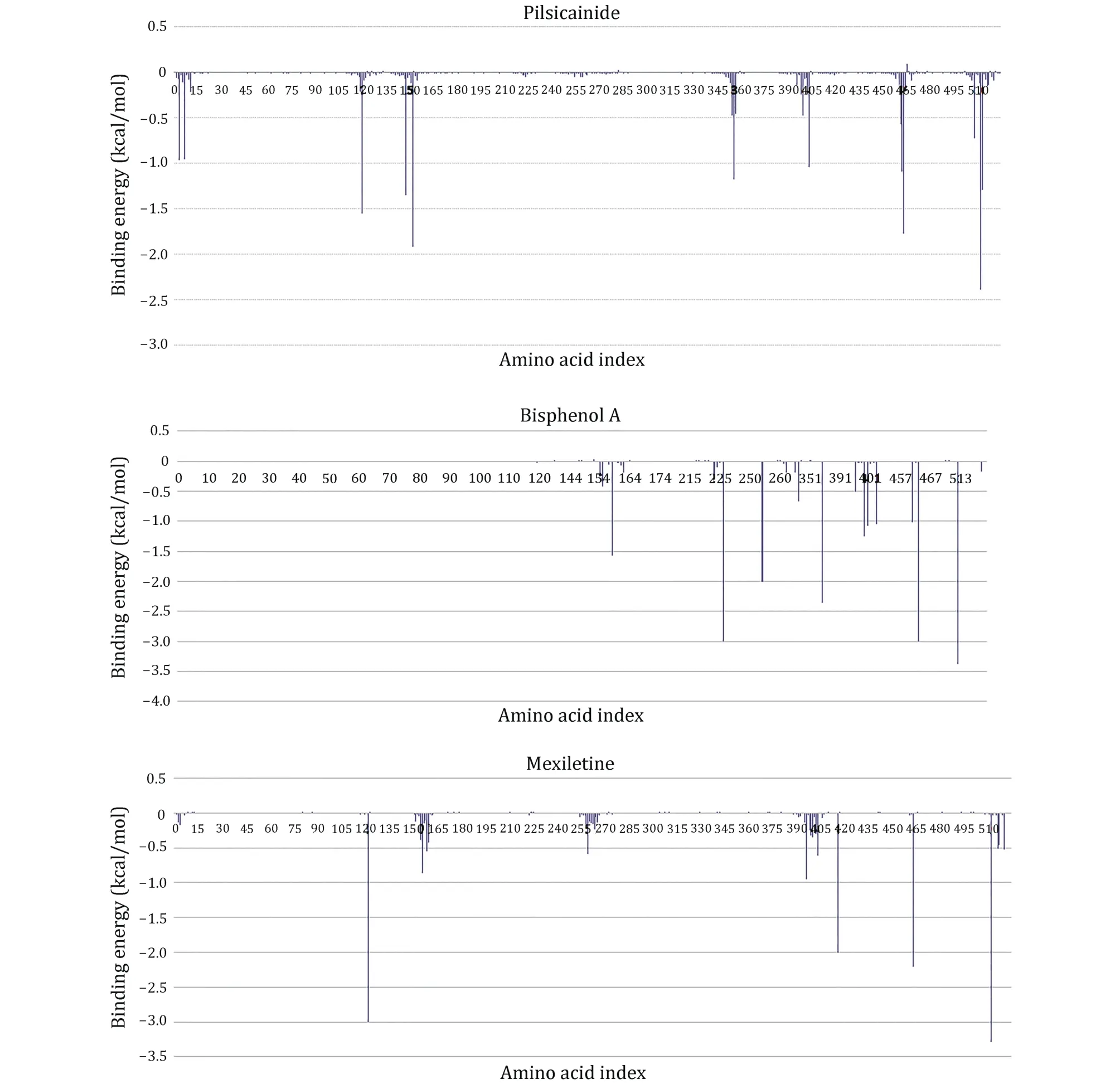
Fig.6 Residue interaction spectrum between the closed10 and three closed-state LAs.The horizontal axis is amino acid index and the vertical axis is the binding energy
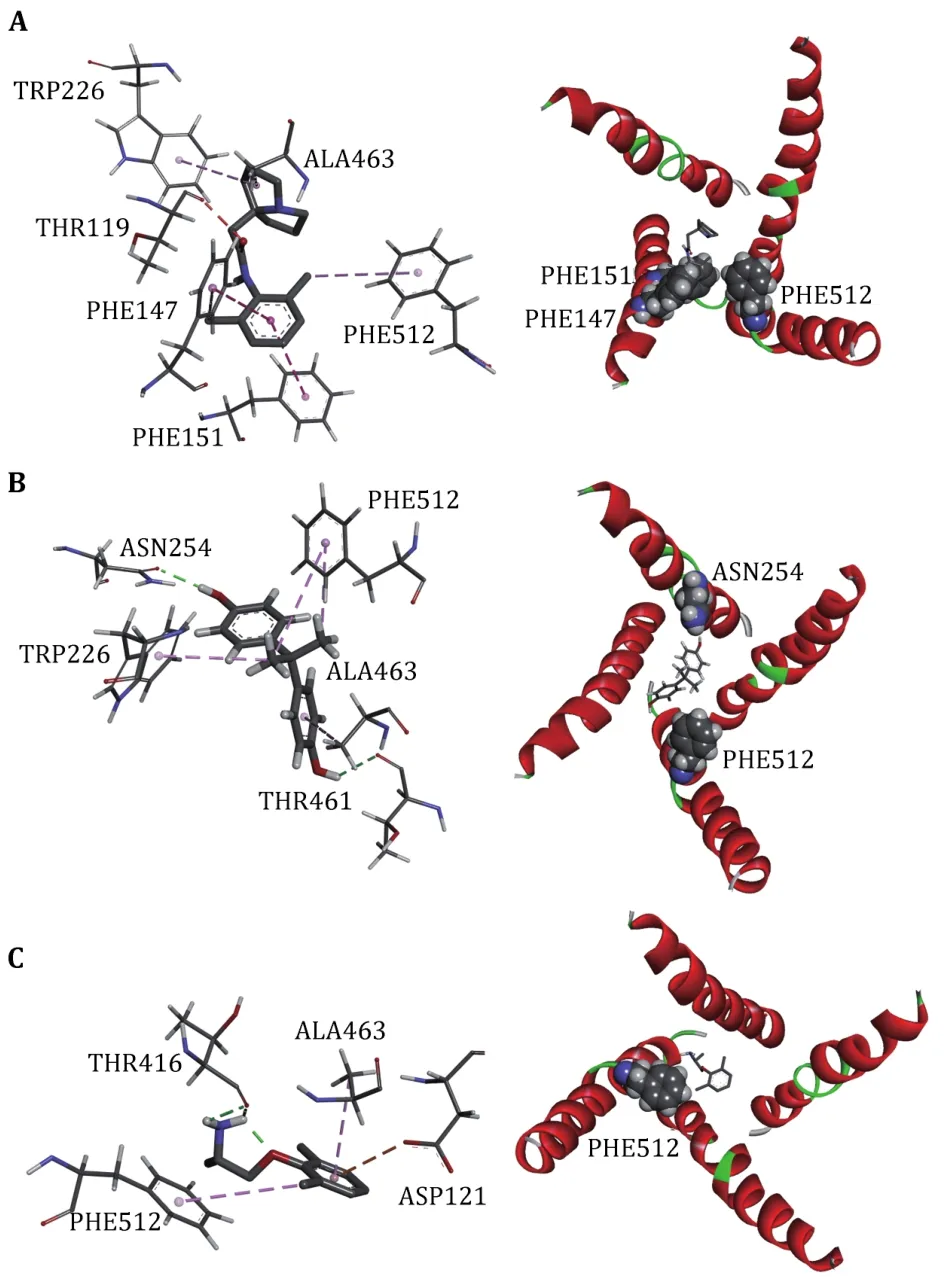
Fig.7 Interactions between Nav1.5 PD and LAs, Pilsicainide (A),Bisphenol A (B) and Mexiletine (C).The interaction diagrams (left column) were calculated and all of the structures were generated by using Discovery Studio client (version 4.1).S6s in four domains (right column) are shown in cartoon and the binding sites are shown in sphere, colored by spectrum
For Bisphenol A, the residues T226, N254, F355, T461,A463 and F512 play major role in the interaction between Bisphenol A and closed10.At the same time, we can see that Bisphenol A interacts with only F512 but not with Y519 (Y1767 in Nav1.5α).Previous researches have shown that Y1767 played a vital role in the use of state-dependent inhibition for LA, and there was no significant effect in the tonic block.F512 is the same binding site of Bisphenol A and Mexletine.
Pilsicainide is the weakest inhibitor among the three closed-state LAs.The main sites of action are T119, F147,F151, W226, A463 and F512.Among these critical sites,only the interaction energy of PHE512 is lower than-2.0 kcal/mol, which contributes the highest binding energy to all residues.Although no experiments have been made to point out whether Pilsicainide has direct interaction with F512, it is an important binding site for LAs.We have reason to speculate that, similar to Bisphenol A and Mexletine, Pilsicainide also interacts with the F512.
CONCLUSION
In this work, the pore domain of voltage gated sodium channel Nav1.5 in closed-state was constructed.The diameter of the final structure in the region of the activation gate is 3.48 Å and is less than the diameter of the hydrated sodium ion.It cannot allow the passage of sodium ions and demonstrates that the model we have built is in closed-state.Furthermore, the interactions between the model and the three closed-state LAs were also studied and the results further validated the reasonability of the built model.Our results may provide a reference for studying the mechanism of interaction between Nav1.5 and LAs and may also provide a theoretical basis for further screening and design of drugs.
METHOD
Prediction of contacts between residues
Recently it was shown that the residue-residue contacts might be determined by co-evolutionary residue pairs,i.e., if two residues in a protein sequence or in two different sequences form direct interaction or contact in 3D structure, they usually co-evolve(Jarzynski 2012; Luntet al.2010; Morcoset al.2011,2014; Weigtet al.2009).DCA calculates a score for each residue pair in a sequence and the score can be used to evaluate if the two residues in the pair can form contact.The detail of DCA has been described in many papers (Jarzynski 2012; Luntet al.2010; Morcoset al.2011, 2014; Weigtet al.2009) and will not be explained here.
DCA uses aligned homologous sequences of the target sequence to analyze the co-evolutionary residues and calculates the score of evolutionary couplings (EC score) between two residues in the target sequence.The sequences of the four domains (DI-DIV) that form the Nav1.5 PD were obtained from NCBI(http://www.ncbi.nlm.nih.gov/) and each of them or each two of them were used as the target sequence(s)to calculate the EC score.The DCA was performed on EVcouplings online server (Aurell and Ekeberg 2012;Hopfet al.2014; Markset al.2011; Morcoset al.2011)and by using a pseudo-likelihood maximization (PLM)approximation (Ekeberget al.2012; Morcoset al.2011).The search tool HHblits (Remmertet al.2011;Vikramet al.2016) was used to generate multiple sequence alignment.In order to assess whether the EC scores can be used to determine residue-residue contacts within and between protein domains, we built an evaluation set.This evaluation set includes the Navchannels with known structures.We calculated the EC scores and distances between each residue pair.The distance of a pair of residues is defined as the shortest atom distance between the two residues.
Structural modeling and selection
Using the ROSETTA-membrane modeling protocol(version 3.4) (Sidharthaet al.2010; Subbotinaet al.2010), the Homology,de novo, and full-atom modeling of the four domains of the PD were built by the crystal structure of NavPaS (PDB ID: 5X0M) as template.Models with lower energy and higher contact coincidence were selected.The energy was calculated by rosettaMP (Alfordet al.2015) and the contact coincidence was defined by the ratio of the predicted contacts and the true contacts of the intra-domains.Then, these selected models were aligned to the corresponding domains of their templates by a structural alignment algorithm TM-align (Zhang and Skolnick 2005) to assemble them to form candidates of the PD structure.The energies of these assembled structures were then evaluated by using rosettaMP and the residue-residue contacts between two domains were analyzed by DCA.Similar to the selection strategy for the monomers or domain structures, the structures with lower energy and higher contact coincidence were selected.Here the contact coincidence was the ratio of the predicted contacts and the true contacts between two domains.The selected structure was kept for further optimization and analysis.
Molecular dynamical simulations and docking
All Molecular dynamical (MD) simulations were done by the software package Amber16 (Caseet al.2016).The setting of parameters and MD process were nearly the same as our previous work (Jiet al.2018) and only briefly described in the following: The modeled structure was oriented by using the PPM server(Lomizeet al.2011) alongZ-axis and then embedded in a homogeneous palmitoyloleoyl phosphatidylcholine(POPC) bilayer and solvated by 20 Å water molecules on both sides of the membranes (Fig.8) by using Charmm-GUI (Leeet al.2015).0.15 mol NaCl was added to neutralize the system.The final system contained 166,700 atoms, including 8316 protein atoms, 444 POPC lipid molecules, 2889 water molecules, 13 Na+ions.After 50,000 steps of optimization (25,000 cycles of steepest descent and 25,000 cycles of conjugate gradient minimization), the system was gradually heated from 0 to 300 K by Langevin thermostat in NVT ensemble.During the equilibration process, the protein was firstly fixed while water and lipid were allowed to equilibrate for 2 ns.Then the contraction of predicted contact residue pairs was restrained by a harmonic potential with force constant reducing gradually from 10 to 0.1 kcal/mol in four 10-ns running steps.Finally, the formal simulations were carried out for a total of 400 ns at constant temperature (300 K) and pressure (1 bar)conditions, which are maintained by using the periodic boundary conditions of NPT ensemble.The module of cpptraj in Amber16 software package was used to analyze the resulting trajectories and the code MOLE 2.0 (Sehnalet al.2013) was used to calculate the pore radius.
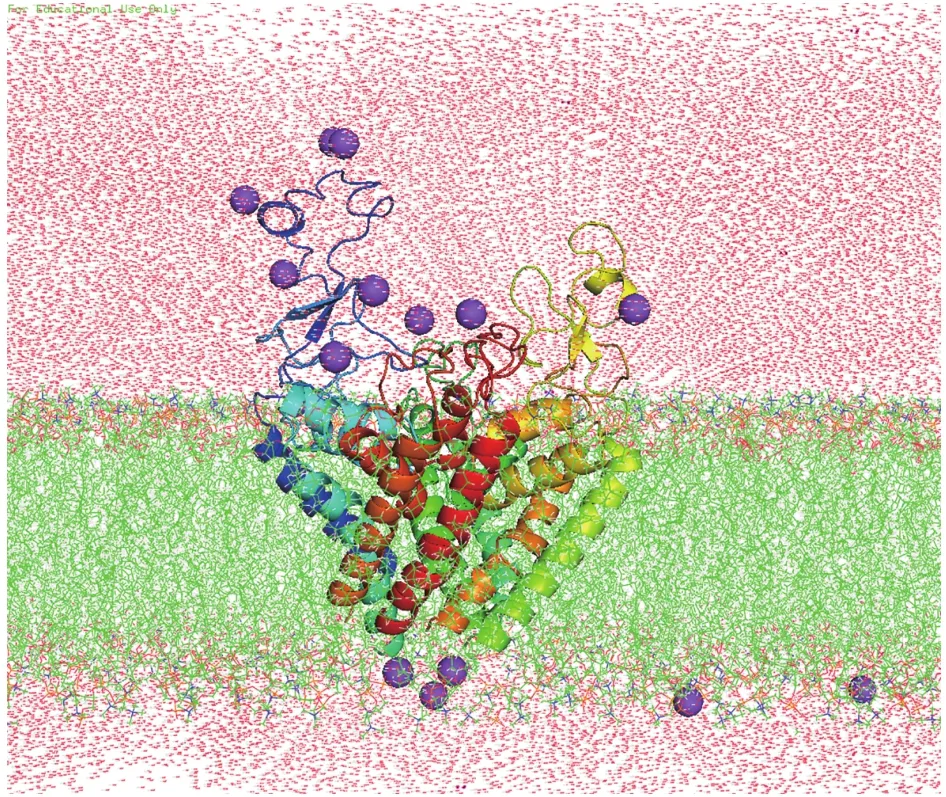
Fig.8 System used for MD simulations of Nav1.5 PD.Nav1.5 PD(in spectrum cartoon) embedded in a POPC lipid bilayer (in green stick).Sodium ions are purple
For the docking calculations, three closed-state LA drugs (Pilsicainide, Bisphenol A and Mexiletine) were downloaded from the ChemSpider (http://www.chemspider.com/).These compounds are sorted in supplementary Table S2 according to theirKdvalues.In this work, Autodock4.2 was employed for docking calculations and Autodock Tools (ADT) (Morriset al.2009) was used for preparing molecules and all of the hydrogens were added by using REDUCE (Wordet al.1999).The docking protocol is similar with the previous literature on ligand docking to Navchannels(Lorenaet al.2016).Modeled structures were defined as receptors and the coordinates of AutoGrid center were determined by the center of the largest binding pocket, which was generated from receptor cavities.The grid size was set to 100 × 100 × 100 points with grid spacing of 10 Å.The grid box included almost the entire channel residues of the receptor.For drug molecules, they were defined as ligands.The procedure was carried out by keeping the protein rigid and the docked ligands were considered flexible.All of the docking decoys were clustered with cutoff 2 according to the root mean square deviation (RMSD) and the lowest-energy docked structures from each cluster were selected as the models of the docked ligand(MDL).Finally, the conformation with the lowest energy was selected as the complex structure for further analysis.
MM-GBSA binding-energy calculations
The selected docking poses of three LA_Nav1.5 complexes were used as the initial structures for MD simulation.Topology and parameter files for ligands were generated using the antechamber program (Wanget al.2006) in the Amber16 package.Partial charges of ligands were calculated using the AM1-BCC method(Araz 2002).Each LA_Nav1.5 complexes was immersed in a 20 Å3-sized cubic box of TIP3P water molecules,and the parameters for amino acids and ligand molecules were assigned using the AMBER-FF14SB force field (Maieret al.2015) and the GAFF force field,respectively.The calculation steps and parameter setup were the same as those for Nav1.5_PD mentioned above.For each LA_Nav1.5 system, binding energies were computed using the popular Molecular mechanics/Generalized Born Surface Area (MM-GBSA)module of AMBER after equilibrium and using a 1-ps frame separation interval.
According to the MM-GBSA protocol, the free binding energy estimation (ΔGbind) can be calculated as follow:
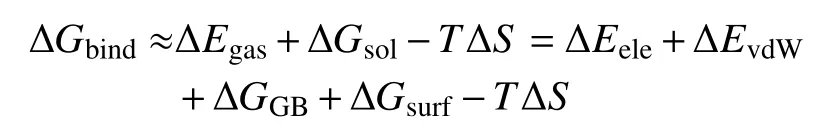
In the above equation, ΔGbindis decomposed into different energy terms.Because the structures of LA_Nav1.5 complex, receptor Nav1.5, and ligand LAs are extracted from the same trajectory, the internal energy change is canceled.Thus, the gas phase interaction energy (ΔEgas) between Nav1.5 and LA is the sum of electrostatic binding energies (ΔEele) and the estimate of the lipophilic van der Waals interaction energy (ΔEvdW)between the Nav1.5 and LA.The solvation free energy(ΔGsol) is formed by the polar and non-polar energy terms.The polar solvation energy (ΔGGB) was calculated by using GB model.The non-polar contribution was calculated based on the solvent accessible surface area(ΔGsurf).
AcknowledgementsThis work is supported by the Central Public-interest Scientific Institution Basal Research Fund, YSFRI,CAFS (20603022019023 and 20603022017006) and The Independent innovation and transformation of achievements of Zaozhuang (2019GH01).
Compliance with Ethical Standards
Conflict of interestXiaofeng Ji, Yanzhao Huang and Jun Sheng declare that they have no conflict of interest.
Human and animal rights and informed consentThis article does not contain any studies with human or animal subjects performed by any of the authors.
Open AccessThis article is licensed under a Creative Commons Attribution 4.0 International License, which permits use,sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material.If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.

- Biophysics Reports的其它文章
- Adaptive optics in super-resolution microscopy
- Ferroptosis and its emerging role in tumor
- An efficient method for the site-specific 99mTc labeling of nanobody
- Instant multicolor super-resolution microscopy with deep convolutional neural network
- PN-ImTLSM facilitates high-throughput low background single-molecule localization microscopy deep in the cell
- Mutual interaction of microbiota and host immunity during health and diseases