
基于单样本特异性的疾病亚型识别方法研究
2021-12-29 03:55:26田显阳季松雨张堃杜映璇张媛媛
现代计算机 2021年31期
田显阳,季松雨,张堃,杜映璇,张媛媛
(青岛理工大学信息与控制工程学院,青岛 266520)
0 引言
癌症指的是细胞不正常增生,且这些增生的细胞可能侵犯身体的其他部分,是控制细胞分裂增殖机制失常而引起的疾病。癌细胞除了分裂失控外,还会局部侵入周遭正常组织甚至经由体内循环系统或淋巴系统转移到身体其他部分。正是癌症的复杂致病机理导致了癌症具有高度异质性,这种异质性使得同一种癌症在临床上常常被分为不同的类型,即癌症亚型,每个亚型往往具有不同的生物标记物。由于癌症亚型在基因上存在差异,因此相同的药物与方法用于治疗的效果不同。基于癌症亚型的分类,临床医生可以指定精准的治疗方案,评估患者的预后。因此,利用组学数据实现对癌症的精准识别,在临床上有极其重要的作用。另一方面,由于癌症异质性的显著存在,不同患者之间在疾病进展、临床疗效、放化疗敏感性及预后等方面差异巨大,深入探讨癌症分子生物学特征及其与临床表现、放化疗敏感性的相关性,从传统形态学分型转变到分子分型,实现从“异病同治”到“同病异治”的转变,有利于对癌症的精准诊断、预后分层、肿瘤分期、指导治疗、复发监控及药物研发[1]。癌症亚型是指在癌症起源处具有相似分子机制的样本群,分子机制通过亚型特异性的突变和表达特征得到反映[1-2]。通过基因组学、转录组学、表观基因组、蛋白质组学、代谢组学等技术对癌症进行不同层次的研究,以数据驱动的方式挖掘生物信息进而进行癌症亚型识别具有重要意义[3-4]。
本文基于P-SSN方法[11],利用基因表达数据,考虑样本特异性信息,以正常样本作为参考矩阵构建参考网络,并将每个癌症样本分别添加到参考矩阵中构建扰动网络,进而构建单样本下的差异基因网络;其次,基于单样本网络,构建信息边缘矩阵,并利用信息熵提取样本网络的边特征,最后构建样本-样本相似性矩阵(图1);最后,利用层次聚类对样本进行聚类,进而确定癌症的亚型。通过与原始特征进行聚类的结果比较,我们发现从网络角度衡量样本的相似性具有明显优势。
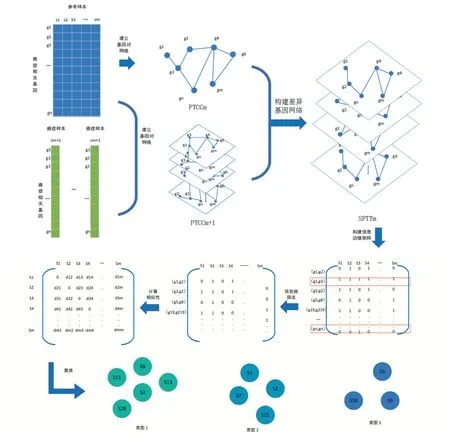
图1 基于单样本网络识别癌症亚型框图
1 材料与方法
1.1 数据来源及预处理
为了系统地分析方法在癌症亚型识别方面的优势,本文从美国生物信息技术中心NCBI(www.ncbi.nlm.nih.gov)中的GEO数据库中下载骨髓增生异常综合征(MDS)转化为急性髓细胞性白血病(AML)的基因表达数据(GSE15061)。该数据包含MDS、AML和正常样本三类。该数据中大多数基因在样本中都存在相似性或是无关性,仅有极少数的基因具有表达值的特征即为显著差异基因,因此原始数据不具备直接用于后续研究与分析的价值与意义,而且将此类基因保留定会增加后续的工作量,加大建立患者基因网络建立的难度,因此需要基因差异表达分析[12],选择具有显著意义的基因构建网络。
本文使用了NetworkAnalyst对基因样本数据进行了基因差异表达分析。NetworkAnalyst是一个基因表达谱和荟萃分析的可视化在线分析平台,集成了先进的统计方法和创新的数据可视化系统,可以进行差异分析和对差异分析结果进行功能分析和网络分析。本文使用了筛选后包含250例样本,其中包括了实验组:100例表现型为AML的癌症患病样本,100例表现型为M DS得到癌症患病样本;对照组:50例不患此癌症的样本。本文使用的差异分析平台是R语言,筛选标准为adjpvalue<0.05和 |log-f old ch ange|>2。从原始数据中总共筛选得到6527个显著差异基因,用于单样本网络的构建。根据Networ kAnalyst对基因数据进行分析与排序获得的差异表达基因结果,从中选取前5000个显著差异基因作为后续的实验数据。
2 方法
本文对癌症基因的亚型识别使用构建基因样本网络的方法,根据对样本网络结构的相似性进行聚类。使用聚类得到的类型与样本原始的亚型标签进行比较,最终依据聚类指标评判构建基因样本网络的方法。具体步骤分为三步,样本特异性基因网络的构建;边缘信息矩阵的构建;聚类分析。
2.1 样本特异性基因网络的构建
样本特异性网络,即单样本网络,是一种基于参考数据集的利用单样本数据构建的生物分子网络,它是一种将复杂网络的理论和方法应用于疾病的研究和药物的开发的方法,可以从系统的角度识别个体疾病所涉及的相互作用或功能失调[11]。
在基因相关性比较中,基因对之间的直接相关系数并不一定可以不受影响地表示基因对间的相关性,因为基因之间的关系很复杂,其往往受到大量其他基因的影响。而偏相关系数可以看做不考虑其他基因影响,单独研究两个基因之间相互关系的密切程度。因此,本文参考P-SSN模型,使用偏相关系数计算基因间的相关性。简单举例,若三个基因相互作用,设为G1,G2,G3。如要分别计算基因对之间的相关性,就不得不考虑第三个基因带来的影响,此时可以运用偏相关系数。G1与G2的偏相关系数可以通过G1与G3线性回归得到的残差R G1,G2与G3线性回归得到的残差RG2来求出G1与G2,的相关系数[13]。
对任意基因X,Y和Z,在正常样本对应的基因表达矩阵,即参考矩阵,中的表达向量记为XT=(x1,x2,…,x m),YT=(y1,y2,…,y m)和ZT=(z1,z2,…,z m),其中m表示正常样本的数量。我们利用偏相关系数,计算在变量Z的影响下,X和Y之间的相关性,计算如下:

GHM分析表明,以乳企为核心的纵向一体化不符合资源配置的规律,导致利益分配向更加不利于奶农的方向发展,养殖者的投资激励不足,退出速度加快。如果我们认为中国的乳制品供给不能完全依靠国际市场、乳业发展带来的就业岗位弥足珍贵,就要扭转以乳企为核心的纵向一体化政策,确立奶农在乳业政策中的核心地位,以保障中国乳业的长远健康发展。改进的思路是将谈判力赋予专用性投资更多的一方,拓展养殖者的外部选择权;同时,要完善竞争机制,在推动“以乳企为核心的纵向一体化”的同时,也给养殖者及其合作社为核心的纵向一体化留出政策空间。
对于每一个癌症样本,将其加入参考矩阵中。类似的,我们可以计算出:

表示该癌症样本对应的特异性基因网络中基因X和Y之间的关系。根据上述步骤,可计算出所有癌症样本对应的单样本显著基因网络,记为:

其中n表示癌症样本的数目。
2.2 边缘信息矩阵的构建
以SS N j(j=1,2,…,n)为基础,将所有单样本特异性网络中的基因对编号,构建边缘信息矩阵,记为M=(m sj)N×n,其中N表示所有网络中的基因对数目,m sj={ }0,1。若S SN j中存在基因对s,则msj=1,否则m s j=0。
为了更好地确定边信息对于癌症亚型的贡献度,我们通过计算每个基因对的信息熵分析了其所提供的信息量,计算公式如下:
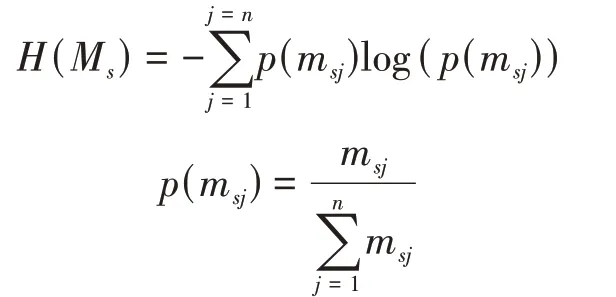
通过上述信息熵公式可知,以-logp(msj)作为信息量,表示事件出现的概率所包含的信息,由于p(m sj)<1,因此以-logp(msj)作为信息量的值必为大于0的值,选择信息熵较小的基因对作为样本特征。同时为了比较不同特征对于癌症亚型识别的有效性,我们计算n个单样本网络S SN j(j=1,2,…,n)中所有基因的度,并将其作为样本的特征。
3 聚类分析
对于不同方法得到的特征,均利用欧式距离计算样本间的距离,进而得到样本相似性矩阵。利用相似性矩阵对样本进行聚类。本文使用层次凝聚聚类法,该聚类法聚类比较快速,能够辨别出干扰信息,并且能够识别出信息,对需要聚类的数据做出相关的划分与处理。为了评估使用不同特征对样本聚类结果的性能,我们使用三个指标进行评价,分别是RI(rand index),ARI(ad⁃justed rand index)和NMI(normalized mutual infor⁃mation)。
用C表示实际的类别划分,K表示聚类结果。定义a为在C中被划分为同一类,在K中被划分为同一簇的实例对数量。定义b为在C中被划分为不同类别,在K中被划分为不同簇的实例对数量。则其中,n表示实例总数,显然,RI的取值范围为[0,1],值越大说明聚类效果越好。但是,RI无法保证随机划分的聚类结果的R I值接近0。
ARI是RI的一个改进版本,总的来说RI是通过计算两个簇之间的相似度来对聚类结果进行评估。而A RI是对R I基于概率正则化的一种改进,其取值范围为[-1,1],A RI取值范围为[-1,1],越高的值表示聚类性能更好。。

NMI通过计算聚类结果和真实结果的互信息,衡量分布的差异性。NMI取值范围为[0,1],值越大说明互相之间的信息量越小,聚类结果越接近于真实结果。它可以通过公式:

计算,其中H(·)代表某一个类的熵值,I(X;Y)代表X和Y的互信息。
4 基于不同特征的聚类结果比较
对于疾病的亚型识别,关键在于基于样本的特征对样本进行聚类,因此,特征的选择直接影响亚型识别的准确性。为了评价构建单样本基因网络后聚类的优点,本文直接使用样本的全部基因表达数据作为对照组的特征(OF,original fea⁃ture),然后使用欧式距离法计算样本相似性,比较两者的聚类指标。其中单样本网络构建后的特征选择又包括三种不同的特征选择方法:单样本网络边缘矩阵(SNE,single sample network edge matrix)、基于信息熵筛选边缘矩阵(EEM,entropy-based edge matrix)和基于单样本网络中基因的度(DSN,degree of genes in single sample net⁃work)作为样本特征。我们对基于信息熵筛选的边缘矩阵作为特征,构建的样本相似性网络,绘制聚类热图(图2),我们发现样本具有明显的聚类特性,从大类上可以分为两大类,与真实的亚型结果相似。
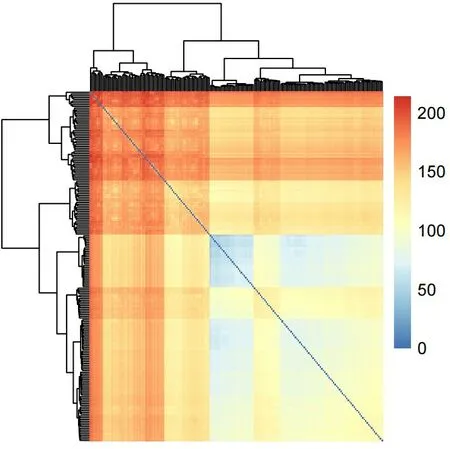
图2 样本的聚类热图
比较四种不同的特征下的聚类结果,如图3所示。通过比较原始特征和基于单样本特异性网络构建的特征,我们发下,无论是基于边缘矩阵,信息熵筛选的边缘矩阵,还是基于网络中节点的度,均比原始特征具有较好的亚型识别性能。原始特征聚类结果较差,主要原因是真正对亚型识别起作用的基因数目往往比较少,是一个全部基因进行计算,引入了太多噪声,稀释了真正的信号。而通过考虑样本的特异性信息,构建单样本网络,从基因间相互作用的系统性角度,可以有效地捕捉到样本的关键信息,对样本的亚型识别具有积极的作用。

图3 不同特征下聚类结果比较
5 结语
本文考虑样本特异性信息,通过构建单样本特异性网络,分析样本网络的特性,从网络角度发现样本间的相似性,提取特异性信息用于样本聚类,进一步识别疾病亚型。通过与使用原始特征进行聚类的结果比较,我们发现,基于单样本特异性网络提取的特征进行聚类的结果与真实亚型的结果更接近。因此,在疾病亚型识别方面,考虑疾病样本的特异性信息将有助于亚型精准识别。
猜你喜欢
家庭医学(下半月)(2020年1期)2020-05-11 02:05:32
海峡姐妹(2018年7期)2018-07-27 02:30:36
特别健康(2018年4期)2018-07-03 00:38:08
特别健康(2018年2期)2018-06-29 06:13:42
中国医疗保险(2017年5期)2017-05-17 08:26:39
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
中国康复理论与实践(2015年10期)2015-12-24 05:42:46
中国病理生理杂志(2015年8期)2015-12-21 12:38:10
现代电生理学杂志(2015年1期)2015-07-18 11:02:16
中国当代医药(2015年30期)2015-03-01 02:08:19
