
配合物Fe(L)2(PF6)2的合成及其对部分过渡金属离子荧光选择
2021-12-17 06:43:26王勇施红
中南民族大学学报(自然科学版) 2021年6期
王勇, 施红
(荆楚理工学院 药物合成与优化湖北省重点实验室, 荆门 448000)
吡啶官能团已经被广泛研究和使用,但仍引起广大科研工作者的兴趣.例如,Kröhnke型吡啶和其他取代的吡啶,在有机和无机结构中都是组成超分子化学的重要部分,具有π-π堆积能力、氢键和配位特性.科学家继续研究这些分子的共轭芳族核产生发光特性的各种应用,如液晶、光敏剂、及生化DNA结合反应机理[1-4].对取代的多吡啶化合物的可能医学应用显示出广阔的前景,归因于它们与金属形成配合物的能力,国际上越来越多的证据表明,能够合成此类取代的吡啶并用于药理学测试.
化合物2,2′:6′,2″-三联吡啶及其衍生物是经典的三齿配体,已在超分子和材料化学中得到广泛应用.由于附加的取代基不仅可以用于修饰配体及其金属配合物的电子性质,还可以通过进一步的衍生化反应引入新的官能团[5-7].在过去的几十年中,科学家已经对d6过渡金属的多吡啶配合物在光致设备中的潜力进行了深入研究.其中较长的激发态寿命和足够高的量子产率是进一步光诱导过程的基础,这些过程在光致发光中起着重要的作用.二价铁的多吡啶配合物由于其光物理、光化学和电化学性质的独特结合而被认为是光分子器件设计的标志性构建基体.此外,这些性质通常在很大程度上可以通过分子大小、形状、拓扑和电子效应进行调节[8-10].
本文以4′位置取代的三联吡啶配体,4′-苯基-2,2′:6′,2″-三联吡啶为原料设计合成一个含三联吡啶配合物Fe(L)2(PF6)2(L=4′-苯基-2,2′:6′,2″-三联吡啶),并对该化合物进行红外、电子吸收光谱、元素分析和X射线单晶衍射等结构表征和谱学性质研究,利用荧光光谱法进一步研究了该化合物对部分重金属离子Fe(Ⅲ)、Pb(Ⅱ)、Co(Ⅱ)、Cr(Ⅲ)、Cd(Ⅱ)、Fe(Ⅱ)、Mn(Ⅱ)、Ni(Ⅱ)、Zn(Ⅱ)的荧光响应.
1 实验部分
1.1 仪器与试剂
溴化钾(光谱纯)、99%硫酸亚铁铵(Alfa Aesar);无水乙醚、六氟磷酸铵、乙腈、二氯甲烷(分析纯,国药集团化学试剂);配体L(L = 4′-苯基-2,2′:6′,2″-三联吡啶)参照文献[11]制得.
电子吸收光谱仪(JACSO V-770型,日本JACSO);荧光分光光度计(F-4600,日本日立);元素分析仪(Vario EL cube,德国元素);小分子单晶衍射仪(Saturn724+,日本Rigaku);傅里叶红外光谱仪(Nicolet iS50 型,美国赛默飞世尔);核磁共振波谱仪(400 M`Hz, AVANCE Ⅲ TM HD,瑞士Bruker).
1.2 配合物Fe(L)2(PF6)2⋅CH3CN·H2O(1)的合成
将两当量L配体(324 mg, 1.0 mmol)的二氯甲烷溶液(25 mL)加入到一当量硫酸亚铁铵(324 mg, 1.0 mmol)的水溶液(25 mL)中, 室温反应5 h,之后向其中加入六氟磷酸铵(170 mg, 1.1 mmol),立即有蓝色沉淀出现,继续反应1 h.将该蓝色混合物过滤后,分别用水(3×30 mL)、2-丙醇(2×25 mL)和二乙基醚(2×20 mL)洗涤,空气中晾干,可得到蓝色固体目标产物.将该蓝色固固体在乙醚-乙腈(V∶V= 30 mL∶10 mL)的混合溶液中重结晶,可以得到纯的深蓝色目标配合物Fe(L)2(PF6)2,184.2 mg, 36%.分子式C44H35F12FeN7OP2,元素分析的理论值:C 51.63, H 3.45, N 9.58;实验值:C 51.36, H 3.34, N 9.39.红外(KBr压片,cm-1):3237, 3102, 3059, 3036, 3001(Ar—H伸缩振动), 1685, 1653, 1635, 1615, 1576, 1519, 1560, 1541, 1522, 1507, 1497, 1487, 1467, 1457, 1450, 1435(芳环Ar—CC—骨架伸缩振动), 1419, 1387, 1374, 1363, 1338, 1284, 1247, 1165, 1111, 1085, 1034, 893, 876, 793, 764, 733, 720(Ar—C—H面外弯曲振动), 681, 669, 655, 617, 532, 522, 510.1H NMR(400 MHz, CDCl3),δ: 9.69(2H, s, Ar—H), 9.09~9.07(2H, d,J=8 Hz), 8.57~8.55(2H, d,J=7.44 Hz),8.07~8.03(2H, td,J=1.08 Hz, 7.92 Hz), 7.86~7.82(2H, t,J=7.58 Hz), 7.75~7.72(1H, t,J=7.38 Hz), 7.30~7.28(2H, d,J=5.04 Hz), 7.22~7.18(2H, t,J=6.46 Hz).电子吸收光谱,λmax/nm[ε/(dm3·mol-1·cm-1)]: 201(121275), 243(38838), 275(74102), 285(99098), 320(57413), 364(8983), 565(29654).
1.3 单晶结构测定
将尺寸合适的X射线衍射分析配合物的单晶安装在玻璃棒上,并在CCD衍射仪上采用石墨单色器MoKα靶(λ= 0.71073 Å )在123 K的条件下使用ω扫描模式收集.原始数据用Crystal Clear程序进行经验吸收校正、优化和还原,通过直接方法解析结构,并使用傅立叶差分技术通过SHELXL-2016/4[12]程序解析.除无序原子外,所有非氢原子均用各向异性位移系数进行精修,同时通过理论加氢原子作为固定成分,参数通过SHELXL-2016程序进行全矩阵最小二乘法修正.氢原子均使用各向同性位移系数U(H)= 1.2U(C)或1.5U(C),并且使用SHELXL-2016/4允许它们的坐标加在它们各自的碳上.表1和表2分别总结了晶体参数、数据收集和提纯的详细信息.表1总结了化合物的晶体学参数、数据收集、结构测定和精修信息,表2列举了部分化合物的重要键长和键角.

表1 配合物的晶体学数据收集、结构测定和精修的详细信息Tab.1 Detail of the crystallographic data collection, structural determination and refinement for the complex

表2 配合物的部分键长(Å)和键角(o)Tab.2 Selected bond length(Å) and bond angle(°) for the complex
2 结果与讨论
2.1 合成与表征
配体L参照文献合成,其核磁共振氢谱如图1所示. 将两当量L配体的二氯甲烷溶液与一当量硫酸亚铁铵的水溶液室温反应,在六氟磷酸铵存在的条件下直接反应制得目标配合物Fe(L)2(PF6)2,对于该配合物采用重结晶法进行纯化,之后通过元素分析、电子吸收光谱、核磁共振氢谱(图2)、红外光谱和X-射线单晶衍射分析等分析测试手段对其进行结构表征确认.

图1 配体L核磁共振氢谱Fig.1 IH NMR of ligand L
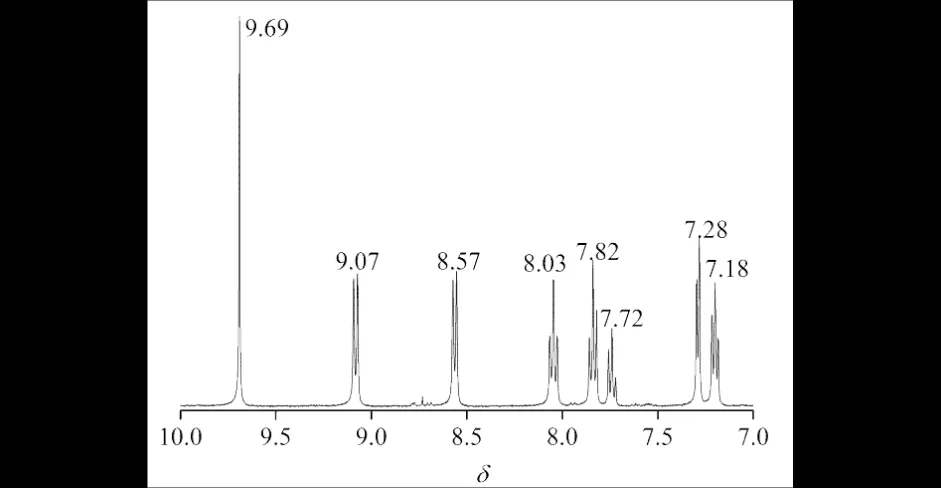
图2 配合物的核磁共振氢谱Fig.2 IH NMR of the complex
配合物在3102、3059、3036和3001 cm-1为芳香环C—H伸缩振动峰,1685、1653、1635、1615、1576、1519、1560、1541、1522、1507、1497、1487、1467、1457、1450和1435 cm-1为芳香环Ar—CC—骨架伸缩振动, 793、764、733和720 cm-1为芳香环C—H面外弯曲振动.
2.2 晶体结构
配合物晶体结构为单斜晶系P21/n空间群,其最小不对称单元包含一个[Fe(L)]2+阳离子、两个PF6-平衡阴离子、一个未配位的乙腈溶剂和一个未配位的水溶剂分子.配合物的阳离子结构如图3所示.配合物是一个扭曲的八面体配位环境,中心铁采用扭曲的[FeN6]八面体六配位,被来自两个配体L的六个氮原子占据.
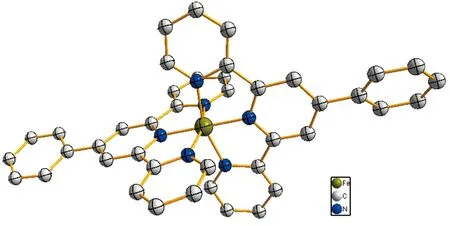
图3 配合物的阳离子结构图Fig.3 Cationic structure diagram of the complex
配合物的轴向键长[Fe—N(2)(1.880(3) Å)和Fe—N(5)(1.874(3) Å)]明显短于径向键长[Fe—N(1)(1.988(3) Å), Fe—N(3)(1.978(3) Å), Fe—N(4)(1.973(4) Å) 和Fe—N(6)(1.965(4) Å)],这与文献报道的化合物[Fe(tpy)2]Cl2[13]比较类似.两个tpy部分之间的二面角为91.6°,这表明两个配体彼此基本垂直.tpy部分与苯基之间的扭转角分别为30.6o和15.1o.Fe⋅⋅⋅Fe之间的最短分子间距离为9.247 Å.两个平行的多吡啶配体基团面对面形成偏移,两个分子之间的多吡啶配体的距离足够短,配合物中芳香环之间最短垂直距离为3.38 Å(图4),在π-π堆积距离(约3.4~3.6Å)[14-15]之间,这表明配合物可能存在一定的分子间相互作用.
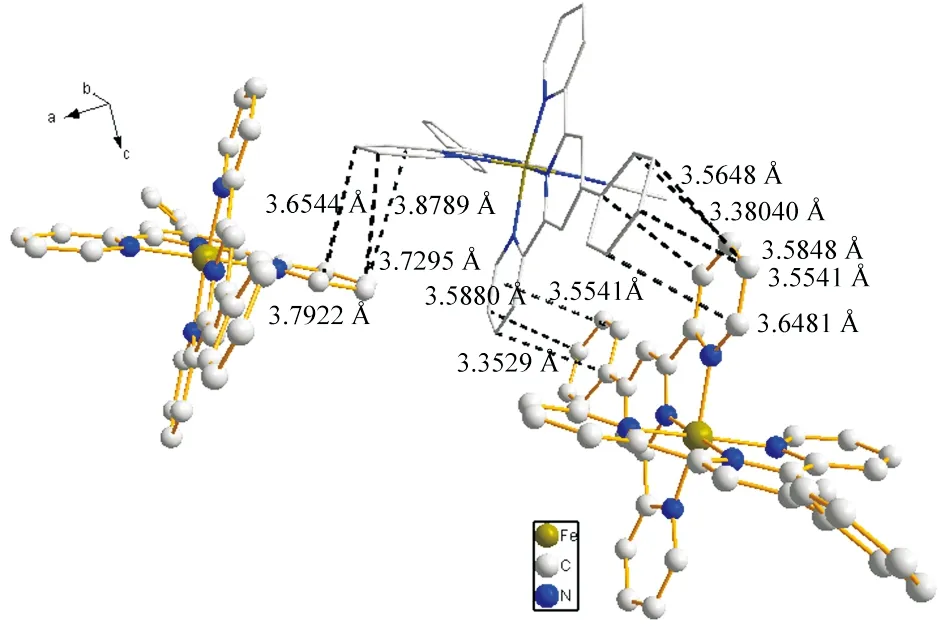
图4 配合物的分子堆积图Fig.4 Molecular stacking diagram of the complex
2.3 电子吸收光谱
配合物的电子吸收光谱是在乙腈溶液中室温测试,浓度为1.0 × 10-5mol·dm-3,其谱图见图5,相关实验数据见实验部分.为了研究配合物的电子吸收光谱,在相同浓度测试了其前体L和(NH4)2Fe(SO4)2的电子吸收光谱.如图5所示,配合物在201、243、275、285、320和364 nm 处的电子吸收光谱主要来自于配体L,可以归属为π→π*跃迁.这些吸收带是具有吡啶环的化合物的特征吸收谱[16-17].相对于前体,配合物在565 nm出现的新峰可以归属为MLCT(metal-to-ligand charge transfer) 跃迁,这与文献报道是比较吻合的.
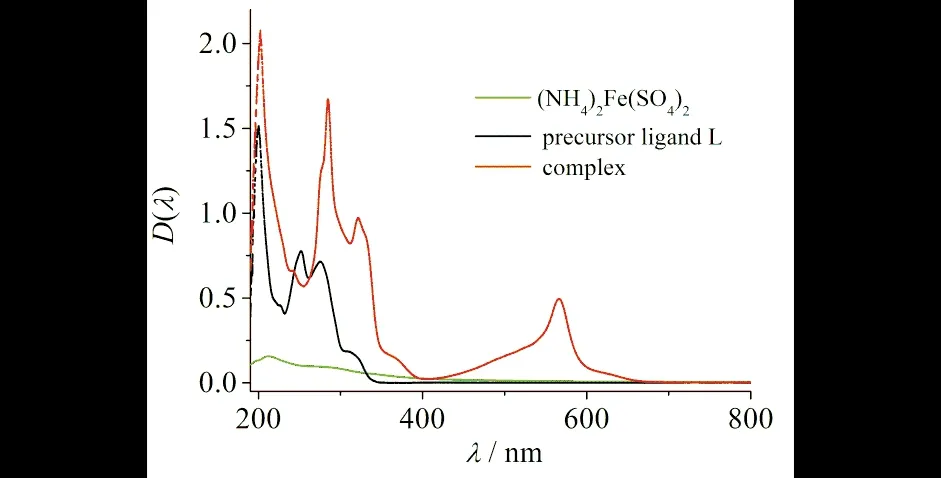
图5 配合物及其前体L的电子吸收光谱Fig.5 Electronic absorption spectra of the complex and its precursor ligand L
2.4 荧光光谱
为了解配合物的荧光性质和荧光的来源,研究了配合物发射光谱,并测试了其前体L的荧光光谱.荧光测试条件:最大发射波长为311.0 nm,最大激发波长为279.0 nm,狭缝宽度(发射光谱为5.0 nm,激发光谱为2.5 nm),PMT电压为700 V,扫描速率为2400 nm·min-1;溶剂:CH3CN(分析纯),浓度为1.0 × 10-5mol·dm-3.配合物在279 nm激发时,在311 nm出现最大发射波长,这可以归结为二价铁离子到4′-苯基-2,2′:6′,2″-三联吡啶配体的金属-配体电荷转移(MLCT).与配体L相比,配合物在311 nm的发射强度相对于配体L强度显著增强,而在354 nm峰几乎完全消失(图6).
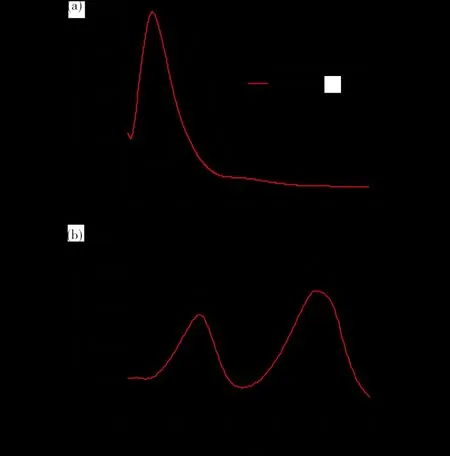
图6 配合物及其前体L的荧光发射和激发光谱Fig.6 Emission and excitation spectra of the complexand its precursor ligand L(a) 荧光发射光谱;(b) 激发光谱
研究了各种常见重金属离子Fe(Ⅲ)、Pb(Ⅱ)、Co(Ⅱ)、Cr(Ⅲ)、Cd(Ⅱ)、Fe(Ⅱ)、Mn(Ⅱ)、Ni(Ⅱ)、Zn(Ⅱ)加入对化合物荧光选择识别作用和发射强度的影响,具体如下:将配合物配成10-5mol·dm-3的CH2Cl2-CH3CN(V∶V=1∶24)溶液,将Cu(OAc)2·H2O配成10-2mol·dm-3的CH3CN-H2O(V∶V=24∶1)溶液,之后在石英比色皿中加入3 mL配合物,3 μL Cu(OAc)2·H2O溶液,确保化合物1与Cu(Ⅱ)离子二者摩尔比为1∶1.接着保持配合物物质的量不变,向其中逐渐滴加Cu(Ⅱ)离子进行荧光发射光谱扫描.其他盐PbCl2、Co(OAc)2·H2O、Fe(NO3)3·9H2O、Cr(NO3)3·6H2O、Cd(NO3)2·4H2O、ZnCl2·6H2O、(NH4)2Fe(SO4)2·6H2O、Mn(OAc)2·4H2O、NiCl2的荧光选择识别实验操作与Cu(Ⅱ)的荧光扫描光谱方法类似.
随着Zn(Ⅱ)离子的加入,配合物的荧光强度逐渐增强,增加到一定程度不再改变;随着Cd(Ⅱ)离子的加入,配合物的荧光强度增强,当加到2 μmol·L-1时,化合物的荧光有所降低,但是最终荧光强度仍然强于配合物.配合物的荧光强度随着各种不同金属离子,如Fe(Ⅲ)、Pb(Ⅱ)、Co(Ⅱ)、Cr(Ⅲ)、Fe(Ⅱ)、Ni(Ⅱ)和Fe(Ⅱ)的加入逐渐减弱,最后配合物的荧光强度几乎完全猝灭.对于Mn(Ⅱ)离子,随着金属离子的加入,配合物的荧光强度有所降低,降低到一定程度,其荧光强度不再发生变化.
配合物对部分金属荧光离子检测相关数据见表3.从表3可知:金属离子Cd(Ⅱ)和Zn(Ⅱ)离子对配合物具有荧光增强的效应,其中Zn(Ⅱ)离子荧光增强效应强于Cd(Ⅱ),这可能是由于Zn(Ⅱ)离子相对与Cd(Ⅱ)有较小的离子半径,更容易与配合物发生相互作用.对于Fe(Ⅲ)、Pb(Ⅱ)、Co(Ⅱ)、Cr(Ⅲ)、Fe(Ⅱ)、Mn(Ⅱ)、Ni(Ⅱ)和Fe(Ⅱ),都对配合物具有荧光猝灭效应,其中Co(Ⅱ)、Fe(Ⅲ)和Cr(Ⅲ)对化合物的选择性最好.

表3 配合物对部分金属荧光离子检测相关数据Tab.3 Related data of selected metal ion fluorescence detection for the complex
3 结语
以硫酸亚铁铵和配体L为原料,在NH4PF6存在的情况下直接反应制得含三联吡啶配合物Fe(L)2(PF6)2,应用元素分析、电子吸收光谱、红外光谱和X射线单晶衍射分析等技术对配合物进行了结构表征.并研究了部分金属离子对配合物的荧光选择性,Zn(Ⅱ)和Cd(Ⅱ)增强配合物的荧光发射光谱,其他金属如Fe(Ⅲ)、Pb(Ⅱ)、Co(Ⅱ)、Cr(Ⅲ)、Fe(Ⅱ)、Mn(Ⅱ)、Ni(Ⅱ)和Fe(Ⅱ)等对配合物的荧光发射强度有部分或全部猝灭效应.
猜你喜欢
云南化工(2021年10期)2021-12-21 07:33:28
化工学报(2020年4期)2020-05-28 09:25:24
今日农业(2019年11期)2019-08-13 00:49:02
中国资源综合利用(2017年1期)2018-01-22 02:44:30
山东工业技术(2016年15期)2016-12-01 05:31:08
中国粮油学报(2016年5期)2016-01-23 02:44:53
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:06
郑州大学学报(理学版)(2014年3期)2014-03-01 04:21:05
无机化学学报(2014年12期)2014-02-28 17:33:51
无机化学学报(2014年10期)2014-02-28 17:33:16
