
A practical strategy to subcutaneous administered in-situ gelling co-delivery system of arsenic and retinoic acid for the treatment of acute promyelocytic leukemia
2021-12-17 03:15:58
Department of Pharmaceutics, School of Pharmacy, Fudan University, Shanghai 201023, China
Keywords: Arsenic trioxide All trans retinoic acid Phospholipid phase separation gel Sustained-release Bioavailability Compliance
ABSTRACT Arsenic trioxide (ATO) combined with all trans retinoic acid (ATRA) is the first choice for the treatment of low and medium risk acute promyelocytic leukemia (APL).Clinical studies reported that the combination of ATO and ATRA could achieve a significant curative effect.However,the retinoic acid syndrome,serious drug resistance and the short half-life in vivo which lead to frequent and large dose administration limit the application of ATRA.In addition,the preparations of arsenic are conventional injections and tablets in clinic,which has poor patients’ compliance caused by frequent long-term administration and serious side effects.In order to overcome the above limitations,a phospholipid phase separation gel (PPSG) loaded with ATO and ATRA was developed.ATO+ATRA-PPSG (AAP),as a biodegradable sustained-release delivery system,was the first achievement of co-delivery of hydrophilic ATO and lipophilic ATRA with high drug loading which is the main problem in the application of nano preparation.The prepared PPSG displayed high safety and biocompatibility.The drug in PPSG was released slowly and continuously in vivo and in vitro for up to 10 d,which could reduce the side effects caused by the fluctuation of blood drug concentration and solve the problem of the long treatment cycle and frequent administration.In vivo pharmacokinetics depicted that PPSG could improve the bioavailability,decrease the peak concentration,and prolong the t1/2 of ATO and ATRA.Particularly,AAP significantly inhibited the tumor volume,extended the survival period of tumor-bearing mice,and promoted the differentiation of APL cells into normal cells.Therefore,ATO+ATRA-PPSG not only could co-load hydrophilic ATO and lipophilic ATRA according to the clinical dosage,but also possessed the sustained-release and long-acting treatment effect which was expected to reduce administration time and ameliorate compliance of patients.Thus,it had great potential for clinical transformation and application.
1.Introduction
Acute promyelocytic leukemia (APL),as an aggressive type of acute myeloid leukemia (AML),is caused by the translocation between promyelocytic leukemia (PML) gene on chromosome 15 and retinoic acid receptorα
(RARα
) gene on chromosome 17.The formed PML-RARα
fusion gene expresses PML-RARα
fusion protein that inhibits promyelocytic differentiation,maturation,and apoptosis [1,2].Clinically,arsenic trioxide (ATO) and all trans retinoic acid (ATRA) are the first-line drugs in the treatment of non-high-risk APL [3,4].Although ATO combined with ATRA achieve a high complete remission rate and low cumulative relapse rate [5,6],there are still some side effects and limitations in clinical application including hypoleukemia,cardiac toxicity,disseminated intravascular coagulation (DIC) related to the fluctuation of ATO concentration [7,8],and retinoic acid syndrome,hyperhistamine syndrome caused by long-term and high dosage administration of ATRA [9,10].The apoptosis induced by ATO is time-dependent that the longer the concentration of ATO is maintained in the effective concentration range,the more apoptosis of leukemia cells and better clinical efficacy will be achieved [11].At present,Arsenious Acid and Sodium Chloride Injection or realgar–indigo naturalis formula (RIF) combined with ATRA have been used in the treatment of APL in clinic [2,3].As Zhao et al.reported,a constant ATO concentration of 0.2 mg/l might achieve a safe and effective treatment [12].Arsenious acid and sodium chloride injection,a conventional intravenous preparation,was administered by the continuously slow infusion method that costs 18–20 h to maintain a relatively steady concentration of ATO [13,14].The continuous infusion causes poor patients’ compliance and increases hospitalization expenses [15].In comparison,oral RIF not only possesses a certain curative effect,but is also convenient and cost-effective [16-20].However,the high administration dosage (30 tablets a day) equaled to about 336 mg arsenic,which increases blood drug concentration fluctuation and the risk of adverse reactions [21,22].Whether arsenious acid and sodium chloride injection or RIF,they all have some limitations in application and are necessary to combine with ATRA which is devoted to promoting leukemia cells to differentiate into normal cells [23-26].ATRA oral common preparations,such as tablets and capsules,are mainly applied in clinic.According to the short biological half-life and the dosage of 45 mg/mm[27],patients are required to take about 9 tablets daily and need long-term medication until the symptoms are relieved [2].Despite it has the advantages of high curative effect,no bone marrow suppression,and convenience for administration,long-term and high-dose medication of ATRA inevitably produces serious side effects,including retinoic acid syndrome,renal insufficiency,and hyperbilirubinemia [9,28].In addition,the nature of ATRA,such as water-insoluble,instability and irritation,also limits its clinical application [29].
In order to solve the above problems in clinical application,numerous nano preparations such as liposomes and nanoparticles have been reported [30-41].However,these nanomedicines are still confronted with challenges in clinical transformation,such as low drug loading,complicated preparation processes and difficulty in co-loaded ATO and ATRA.
Herein,to overcome the limitations of both marketing and developing preparations,an ATO and ATRA co-loaded PPSG was developed by one-step vortex method.The prepared ATO+ATRA-PPSG was the first drug delivery system to meet the needs of clinical co-administration.Because PPSG solution is the ethanol solution of phospholipid that could dissolve ATRA and ATO to a large extent,it is not necessary to consider the drug loading which is the main problem in the development of a variety of delivery systems.Thus,the proportion and the dosage of ATO and ATRA could be adjusted flexibly and conveniently according to the clinical therapeutic schedule.In this study,the prepared PPSG solution was injected subcutaneously and formed a phospholipid gel reservoir at the administration site when ethanol diffused to surrounding tissues [42].Rheology,in
vitro
andin
vivo
release behavior and biosafety of AAP were studied.Pharmacokinetics and pharmacodynamic evaluation were performed to investigate the bioavailability of ATO and ATRA and the ability to inhibit tumor growth.2.Materials and methods
2.1.Materials and animals
ATO was obtained from Sinopharm Chemical Reagent Co.,Ltd (Shanghai,China).ATRA was purchased from Sigma-Aldrich (St.Louis,MO,USA).Phosphatidylcholine (PL-100 M) was purchased from Shanghai AVT Pharmaceutical Technology Co.,Ltd.(Shanghai,China).Injection-grade medium chain triglyceride (MCT) was provided by Xinxing Pharmaceutical Co.,Ltd.(Liaoning,China).DIR (1,1-dioctadecyl-3,3,3,3-tetramethylindotricarbocyaineiodide) was purchased from Meilunbio.APC labeled anti-human CD11b was purchased from Thermo Fisher Scientific,Inc.(Waltham,MA,USA).All other chemicals and solvents used were of analytical grade.HL-60 cells (human acute promyelocytic leukemia) were bought from Cell Bank of Shanghai Institute of Biochemistry and Cell Biology (Shanghai,China).The healthy male Sprague-Dawley (SD) rats (160-210 g),male Balb/c mice and male nude mice (18–24 g) were obtained from Shanghai Slac Laboratory Co.,Ltd.(Shanghai,China) and raised in compliance with the guidelines of the Ethics Committee of Animal Center of Fudan University.
2.2.Preparation of blank PPSG and drug loaded PPSG
PPSG was prepared by dissolving 70 mg PL-100 M and 15 mg MCT in 15 mg absolute ethanol under vortex stirring at room temperature for 30 min.The ATO and ATRA were loaded according to the clinical dose ratio (about 1:4).To prepare ATO-PPSG and AAP,the PPSG was firstly prepared according to the above method.ATO which dissolved in 5% sodium hydroxide solution was added into blank PPSG solution (1/1000,w/w),and stirred for 5 min to acquire a homogeneous ATO-PPSG.On this basis,added ATRA into ATO-PPSG (1/250,w/w) and stirred for 15 min to obtain AAP.The drug loading of ATO and ATRA in AAP were 1 mg/ml and 4 mg/ml,respectively.PPSG and AAP solution was sterilized by using 0.22μ
m organic microporous membrane.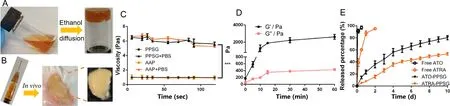
Fig.1–(A) The phase transformation of PPSG in vitro .(B) The phase transformation of PPSG in vivo .(C) Viscosity profiles of PPSG and AAP in solution and gel states (∗∗∗P < 0.001).(D) In vitro release of ATO and ATRA in PBS and PBS containing 2% SDS (pH 7.4) at 37 °C.Data were presented as mean ±SD,n=3.
2.3.Viscosity characterization
The viscosities of PPSG and AAP were measured by a digital viscometer (Gemini 2,Malvern Panalytical Ltd,UK).Added a certain volume of PPSG and AAP on the container to match the size of the rotator,after setting parameters were stable on the screen,recorded the viscosity value.AAP in the dialysis bags were taken out and added onto the parallel plate at 0,5,10,15,30 and 60 min to determine the elastic modulus (G’) and the loss modulus (G").
2.4.In vitro drug release
0.5 ml AAP (containing 0.5 mg ATO and 2 mg ATRA),0.5 ml ATO solution (1 mg/ml) and 0.5 ml ethanol solution of ATRA (4 mg/ml,dissolved in ethanol) were added into the dialysis bags (MWCO=14 kD),respectively.Tied up the two ends of dialysis bag and put it into an EP tube containing 4 ml release medium shaking at 100 rpm at 37 °C.Phosphate buffer (pH 7.4) was the release medium of ATO,and 2% sodium dodecyl sulfate (SDS) contained phosphate buffer was the release medium of ATRA.The release medium was collected and replaced with 4 ml fresh medium at 0.5,1,2,3,4,5,6,7,8,9 and 10 d The ATO and ATRA contents were determined by Atomic fluorescence spectrophotometer (AFS,Ji tian,China) and UV spectrophotometer (at 334 nm).The release kinetic parameters of AAP were elucidated by various mathematical models including the zero order,the first order,Higuchi matrix,Hixson -Crowell,Baker -Lonsdale and Korsmeyer-Peppas release equations.
2.5.Determination of PPSG absorption in vivo
Fluorescence imaging was applied to investigate the absorption of PPSGin
vivo
.The healthy male Balb/c mice were divided into 2 groups (n
=4),and were subcutaneously injected 0.1 ml free DIR solution and DIR-PPSG at DIR concentration of 4 ug/ml.Fluorescence intensity at different time points was obtained by using an IVIS® Spectrum system (PerkinElmer,USA).2.6.Pharmacokinetic Study
Nine male SD rats weighing 250 g were randomly divided into 3 groups (n
=3).After 12 h of fasting,AAP,free ATO and ATRA solution were subcutaneously injected at ATO and ATRA concentration of 2 and 8 mg/kg,respectively.Blood samples (0.5 ml) from retro-orbital vein were withdrawn in heparinized tubes at 0.5 h,4 h,8 h,0.5 d,1 d,2 d,3 d,4 d,5 d,6 d,7 d,8 d and 10 d and then kept frozen at 0 °C until assay.0.2 ml blood sample was added into appropriate amount of concentrated nitric acid and digested in the microwave digester (MDS-8,Sineo,Shanghai) at 150 °C for 10 min and 180 °C for 20 min.The concentration of ATO in blood sample was measured by AFS.At the same time,the plasma was extracted with chloroform and methanol solvents,the content of ATRA was determined by UV (at 334 nm).2.7.In vivo pharmacodynamic study
The heterotopic tumor model of APL was established by secondary inoculation.Firstly,1 ×10HL-60 cells were suspended in 100μ
l Matrigel which was diluted with culture medium and injected into the subcutaneous tissue of the right axilla of male nude mice.When the tumor volume reached about 300 mm,the tumor was separated and homogenized into a uniform single-cell suspension with a glass homogenizer.The single-cell suspension was filtered through a 70 mm cell sieve and added into appropriate amount of red blood cell lysate reagent to remove red blood cells,and then washed with serum-free medium three times.The APL cell suspension was inoculated into the right axilla of male nude mice according to the first transplantation method to build the xenotransplantation model of secondary transplantation.When the tumor size was about 80-100 m,the mice were randomly divided into 5 groups (n
=6) and subcutaneously administrated beside the tumor with 0.1 ml saline,PPSG,free ATO+ATRA (free AA,containing 0.1 mg ATO and 0.4 mg ATRA),ATO-PPSG (containing 0.1 mg ATO) and AAP (containing 0.1 mg ATO and 0.4 mg ATRA) for one-time in total.Body weights and tumor volumes were monitored every 2 d After 18 d of treatment,the tumor-bearing mice were sacrificed to collect serum to analyze biochemical indexes including Alkaline phosphatase (ALT,an indicator of liver function),Alkaline phosphatase (ALP,an indicator in the diagnosis of hepatobiliary diseases) and Creatinine (an detect index the filtration rate of kidney) and collected visceral organs including heart,liver,spleen,lung,and kidney which were fixed by 4% paraformaldehyde for H&E staining,and homogenized the tumor into cell suspension that stained with APC labeled anti-human CD11b for flow cytometry.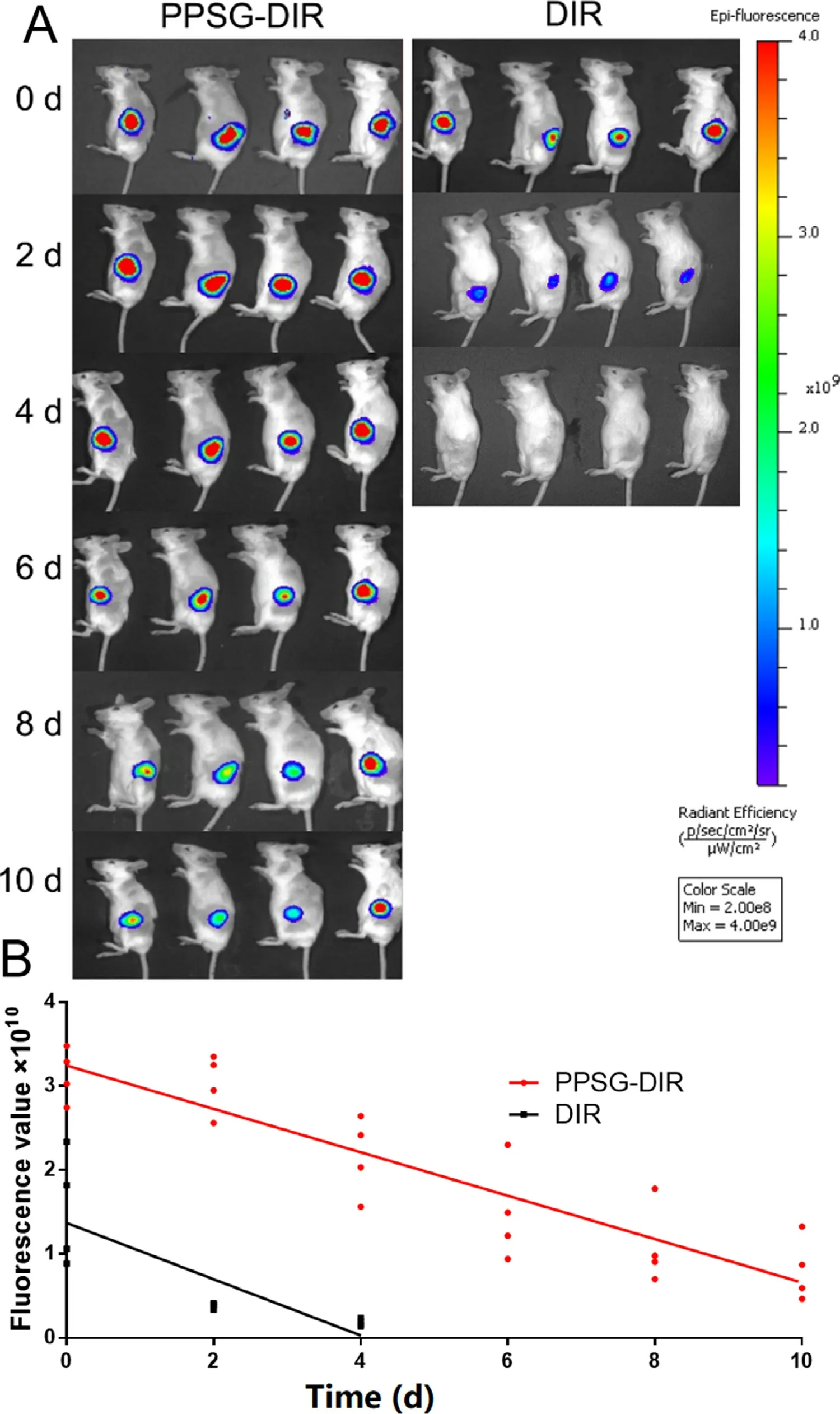
Fig.2–In vivo images of mice after subcutaneous injection of DIR and PPSG-DIR (n=4).
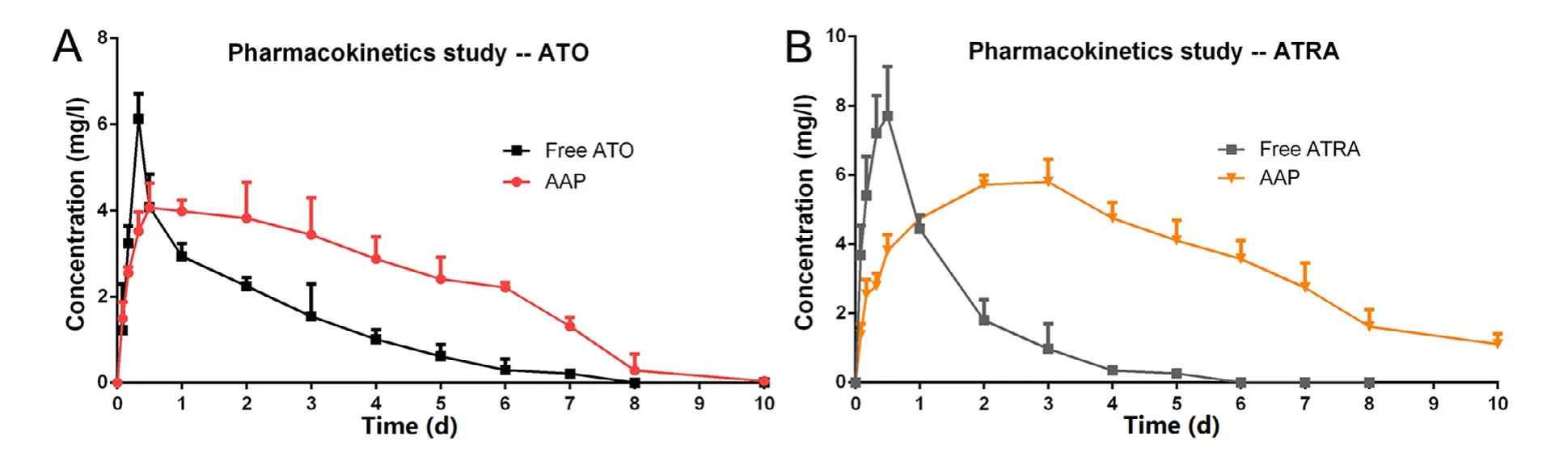
Fig.3–(A) Mean ATO plasma concentration-time curves of the free ATO and AAP.(B) Mean ATRA plasma concentration-time curves of the free ATRA and AAP.(each point represented the mean ±SD,n=3).
2.8.Safety evaluation
Healthy male Balb/c mice were shaved and randomly divided into 4 groups,followed by subcutaneous injection with 0.1 ml saline,PPSG,ATO-PPSG and AAP,respectively.At 1,3,7,14,21 and 30 d post-injection,the skin of the injection site was photographed and the mice were executed to collect skin,heart,liver,spleen,lung,and kidney for H&E analysis.
2.9.Statistical analysis
All the data were expressed as mean ± SD.DAS 3.0 software was applied to analyze the data of the pharmacokinetic study.t-test was applied to determine significant differences by GraphPad Prism 6 (CA,USA).P
value of<
0.05 was significant.3.Results and discussion
3.1.Preparation of PPSG and phase transition
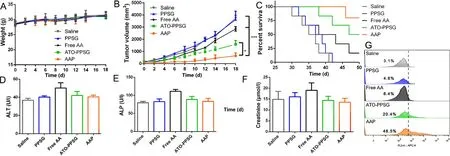
Fig.4–In vivo efficacy of saline,free AA,PPSG,ATO-PPSG and AAP on xenotransplantation model.Body weight (A),tumor volume (B) and percent survival (C) of tumor-bearing nude mice after subcutaneous injection.(∗P < 0.05,∗∗P < 0.01,∗∗∗P < 0.001).Serum biochemical indexes including ALT (D),ALP (E),Creatinine (F) and (G) CD11b + cells percentage of tumor-bearing nude mice at Day 18 after treated with saline,PPSG,free AA,ATO-PPSG and AAP.(each point represented the mean ±SD,n=6).
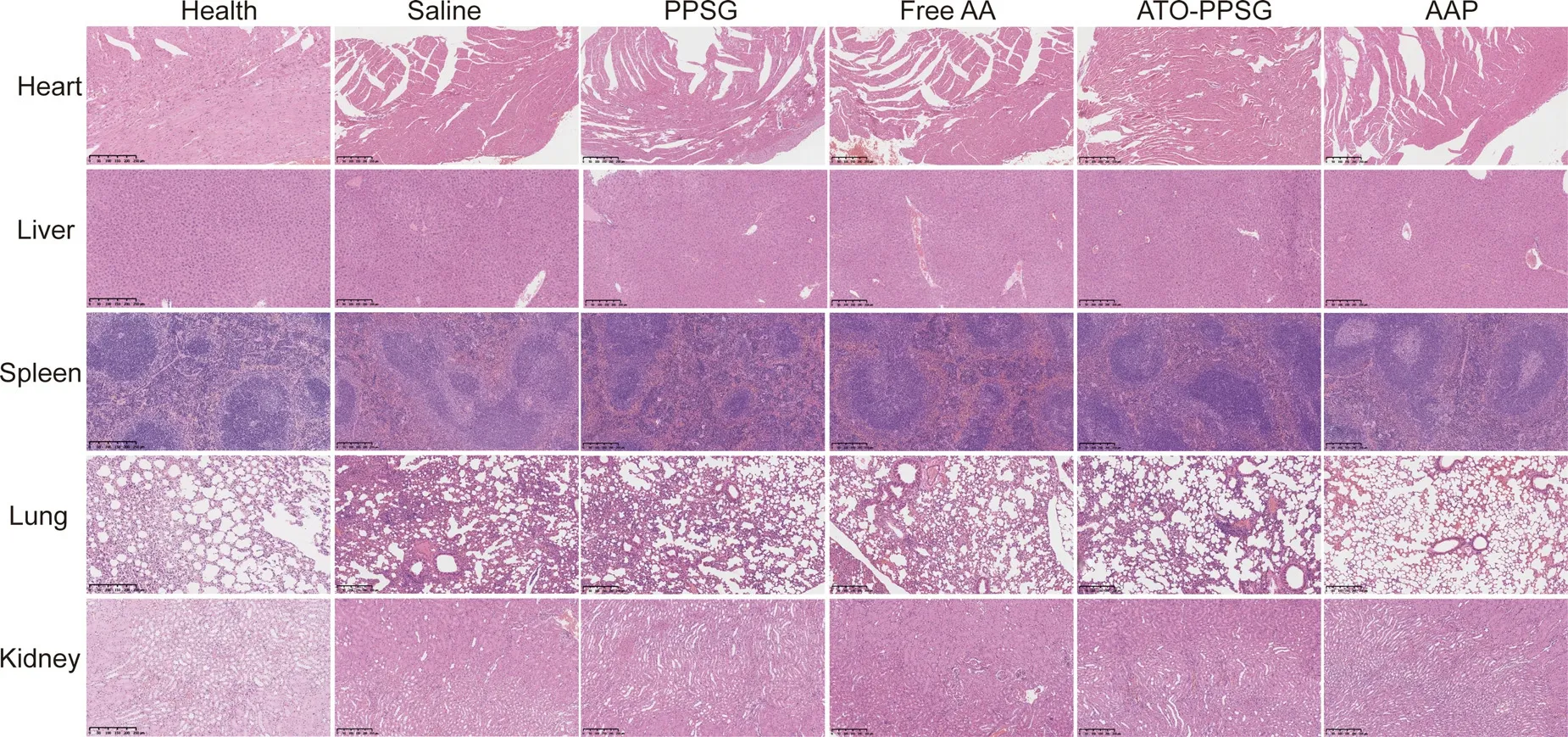
Fig.5–H&E staining of heart,liver,spleen,lung and kidney from each group at Day 18 after subcutaneous injection of saline,PPSG,free AA,ATO-PPSG and AAP,compared with the healthy mouse (scale bar,250 μm).(For interpretation of the REFERENCES to color in this figure legend,the reader is referred to the web version of this article.)
PPSG prepared by vortex method was a transparent homogeneous solution with good fluidity and natural yellow color of phospholipid.After ethanol was replaced by aqueous solutionin
vitro
,the PPSG solution displayed a rapid phase separation and a transition to a semi-solid state (Fig.1 A).Further,in order to investigate the phase transition of PPSGin
vivo
,100 μl PPSG was subcutaneous administration into the right leg of mice,as shown in Fig.1 B,the PPSG solution could form a uniform,soft and smooth gelin
vivo
.The flowability was assessed by measuring the viscosity.As shown in Fig.1 C,the viscosity of PPSG increased significantly after phase transition,which was about 6 times that of PPSG solution (P
<
0.05),suggesting that the immediate transition process of PPSG could establish a stable drug reservoir at administration site.As shown in Fig.1 D,before the phase transition,the values of G’ and G" were about 150 and 55 Pa.With the progress of phase transformation,both G’ and G" increased rapidly over time,and the value of G’ was higher than that of G",indicating that AAP possessed high elasticity.After 15 min,G’ and G" reached a relatively stable stage,suggesting that the sol-gel transformation was completed.And the contents of water and ethanol in AAP gel were about 30.5% and 0.0%.3.2.Drug release in vitro
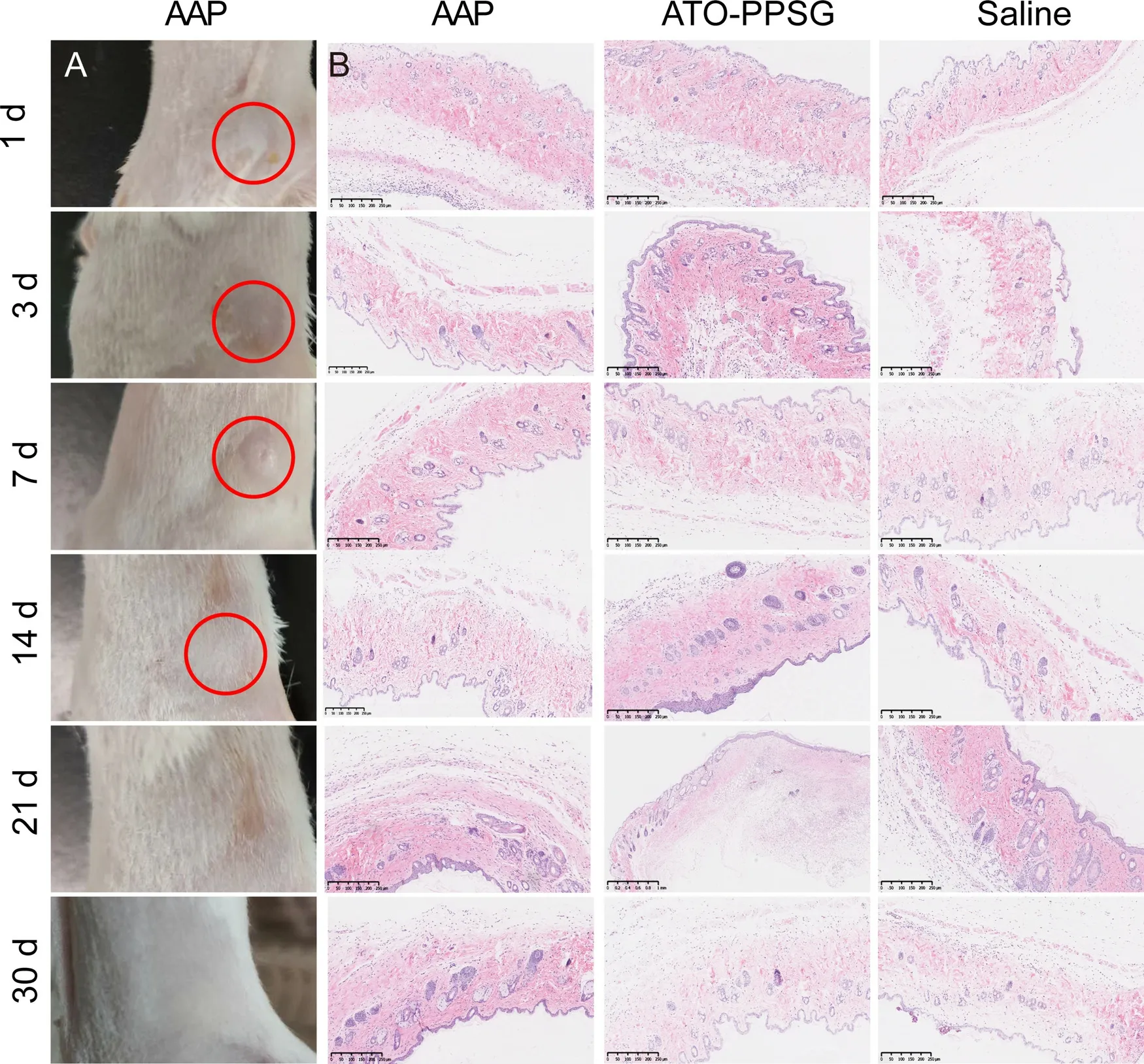
Fig.6–(A) The appearance of the skin at the injection site (red circles).(B) H&E staining of skin from each group at the injection site in 30 d (scale bar,250 μm).(For interpretation of the REFERENCES to colour in this figure legend,the reader is referred to the web version of this article.)
The dialysis method was applied to evaluate the drug releasein
vitro
.As shown in Fig.1 E,ATO and ATRA solution showed a rapid release in PBS and PBS containing 2% SDS,about 90% were released in 2 h and 12 h,respectively.By contrast,the release profiles of ATO and ATRA from AAP displayed a noticeably delayed release of 10 d without initial burst release.30% ATO and 10% ATRA were released within 2 d,80% ATO and 50% ATRA were released within 10 d To clarify the mechanism of drug release,various mathematical models were adopted,including zero-order kinetic model,first-order kinetic model,Higuchi,Hixson-Crowell,Baker-Lonsdale and Korsmeyer-Peppas.As shown in Table 1,thein
vitro
release of ATO from AAP was in good agreement with the first-order model (r
=0.9986),followed by Higuchi model (r
=0.9949),Hixson Crowell model and Korsmeyer-Peppas model (r
=0.9945),demonstrating that the release mechanism might be the result of diffusion.Moreover,the release of ATRA from AAP fit with Baker Lonsdale model (r
=0.9910),followed by Higuchi model (r
=0.9888) and first-order model (r
=0.9844),manifesting that the release was mainly through the dissolution of PPSG which contributed to the sustained release.Therefore,it could be predicted that the slow and continuous release could significantly reduce the fluctuation of blood drug concentrationin
vivo
.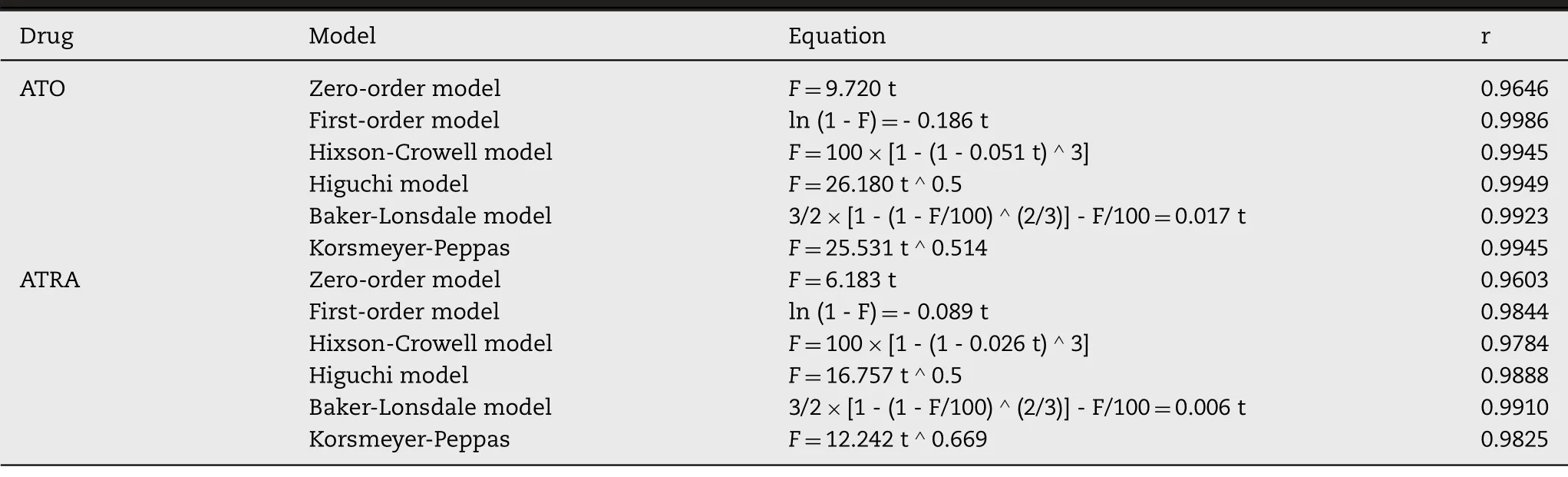
Table 1–The release kinetics of ATO and ATRA of AAP in PBS and PBS containing 2% SDS.
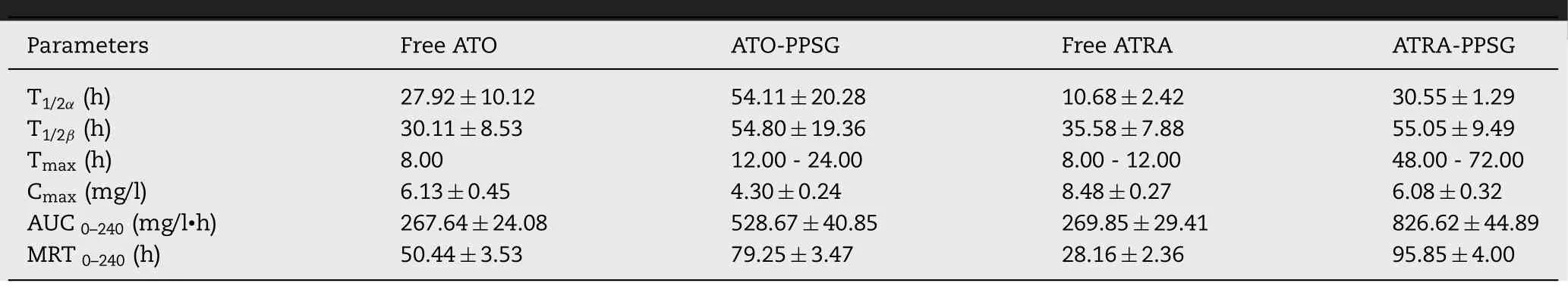
Table 2–Pharmacokinetics parameters of free drug and AAP after subcutaneous administrations in rats (n=3).
3.3.Determination of PPSG absorption in vivo
The absorption characteristics of PPSGin
vivo
were studied by fluorescence imaging.As shown in Fig.2 A&2B,strong fluorescence signals were observed within 10 d in the PPSG-DIR group,and the intensity of the signal decreased slowly over time.In contrast,only a weak signal was observed in the DIR group on the Day 2,and no fluorescence signal was detected on the Day 4.Therefore,the slow absorption of PPSG would possess an excellent sustained-release profilein
vivo
.3.4.Pharmacokinetics study
The pharmacokineticsin
vivo
was investigated by one-time subcutaneous administration in rats.The mean blood concentration-time curves of free drug and AAP were given in Fig.3 and the pharmacokinetics parameters were listed in Table 2 .As shown in Fig.3,compare with free drugs,T
,T
1/2
β
and MRT of AAP increased by approximately 1.5-4.0 times.C
max
decreased by about 30% andT
max
extended by about 28 -50 h,demonstrated that AAP could conspicuously decrease the peak concentration and maintain a relatively constant concentration within 10 d.Importantly,theAUC
of AAP increased by about 3.0 folds that significantly enhanced the bioavailability of ATO and ATRA.Thus,AAP could not only reduce the toxic and side effects caused by the burst release of ATO and high-dose administration for a long term,but also could maintain a relatively stable concentration which was beneficial to decline administration times,improve the therapeutic effect and patients’ compliance.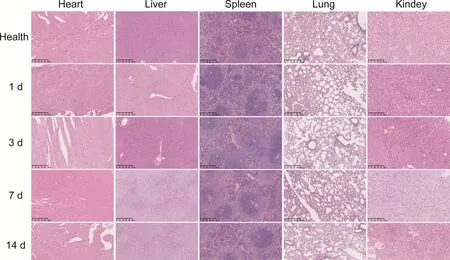
Fig.7–H&E staining of heart,liver,spleen,lung and kidney from each group in 30 d after subcutaneous injection of AAP (scale bar,250 μm).
3.5.Pharmacodynamics study
The xenotransplantation model of APL was established by secondary subcutaneous transplantation of HL-60,and then evaluated the efficacy of ATO-PPSG and AAP in the treatment of APL.When the tumor volume was about 100 mm,0.1 ml saline,free ATO+ATRA (free AA),PPSG,ATO-PPSG and AAP were given subcutaneously for once in total.The body weight of tumor-bearing nude mice in 5 groups remained stable within 18 d (Fig.4 A).As shown in Fig.4 B,the tumor volume grew quickly in saline and PPSG groups,which increased by nearly 20 times on the Day 18 after one-time subcutaneous administration.Free AA inhibited the growth of tumors in the early stage,but the growth gradually became uncontrolled for the rapid absorption of drug solution.By contrast,ATO-PPSG could suppress tumor growth,and the combination of ATRA displayed the best anti-tumor efficacy.The tumor volume of 1500 mmwas applied as the standard to judge the death of mice.According to the survival curve within 50 d (Fig.4 C),the tumor volumes of all mice in saline and PPSG groups were more than 1500 mm,while that of 5 mice in free AA group reached the death standard.In comparison,there were 3 and 1 mice in ATO-PPSG and AAP groups that the tumor volume exceeded 1500 mmin 50 d,respectively.It could be inferred that when subcutaneous administrated the PPSG solution,the immediate phase transition led to form a solid drug storehouse which could release ATO and ATRA slowly and maintain stable blood drug concentration.Therefore,AAP could significantly inhibit tumor growth and enhance the anti-tumor effect.
On the Day 18,the tumor-bearing mice were sacrificed and collected blood,tumors,and visceral organs.Firstly,the serum was separated and the biochemical indexes including ALT,ALP and Creatinine were analyzed.As shown in Fig.4 D-4 F,although all values were in the normal range,the levels of ALT,ALP and Creatinine in ATO-PPSG and AAP groups were inferior to that in free AA group,indicating that encapsulated drugs in PPSG could reduce the hepatorenal toxicity to a certain extent.In addition,the collected tumor was grounded to obtain the tumor cell suspension and then stained with CD11b.As shown in Fig.4 G,the percentage of CD11bcells in AAP group was about 48.5%,which was higher than that in ATO-PPSG group (20.4%),suggesting that the combination of ATO and ATRA could noticeably improve the differentiation ability of APL cells.
To further investigate the pathological changes of tumor-bearing nude mice,histopathological of tissues were observed by H &E staining (Fig.5).No obvious abnormality was found in the sections of heart,liver,spleen,and kidney.But in the section of lung tissue,the alveolar walls in saline,PPSG,free drug and ATO-PPSG groups showed different degrees of thickening.Compared with free AA,there was less thickening in ATO-PPSG group,and almost no thickening in AAP group,demonstrating that AAP could obstruct the proliferation of HL-60 cells and reduce the infiltration of leukemic cells which was consistent with the results of the tumor volume.
3.6.Safety evaluation
To investigate the safety of PPSGin
vivo
,the mice were sacrificed at different time points after subcutaneous administration of AAP,the skin and visceral tissues were collected for HE staining.As shown in Fig.6,there were no swelling and pathological changes at the administration site and surrounding tissues within 30 d HE staining depicted that the skin structure in ATO-PPSG and AAP groups were same to that in saline group.Meanwhile,the visceral tissue structures had no significant change (Fig.7),manifesting that PPSG was a biodegradable,biocompatible and safe controlled release carrier.4.Conclusion
AAP was the first drug delivery system to co-loaded hydrophilic ATO and lipophilic ATRA.The proportion and dosage of ATO and ATRA could be adjusted flexibly in accordance with the clinical treatment plan for the large drug loading.The AAP prepared by vortex method possessed good fluidity and rapid phase transition process.Based on thein
vitro
results,PPSG could control the release of hydrophilic ATO and lipophilic ATRA slowly and persistently for up to 10 d After local administration,the drug reservoir was formed and the absorptionin
vivo
could proof the release characteristics of ATO and ATRAin
vitro
well.The decreased peak concentration,relatively stable blood drug concentration and increasedAUC
value obtained in the pharmacokinetics study could be reasonably inferred that AAP is hopeful to reduce administration times and maintain a constant drug level in the human circulation system.In the pharmacodynamic study,ATO alone or combined with ATRA could accelerate the differentiation of APL cells,inhibit the growth of tumors and prolong survival.Therefore,the AAP with excellent biocompatibility and enhancing bioavailability is promising to improve the medication safety and the compliance of patients,leading to great potential in clinical transformation.Conflicts of interest
The authors declare no conflict of interest.
Acknowledgments
The authors gratefully acknowledge the Science and Technology Commission of Shanghai Municipality (20S11902600).
 Asian Journal of Pharmacentical Sciences2021年5期
Asian Journal of Pharmacentical Sciences2021年5期
- Asian Journal of Pharmacentical Sciences的其它文章
- Spatiotemporally co-delivery of triple therapeutic drugs via HA-coating nanosystems for enhanced immunotherapy
- Investigating the crucial roles of aliphatic tails in disulfide bond-linked docetaxel prodrug nanoassemblies
- Synthetic anti-angiogenic genomic therapeutics for treatment of neovascular age-related macular degeneration
- Enhanced delivery efficiency and sustained release of biopharmaceuticals by complexation-based gel encapsulated coated microneedles:rhIFN α-1b example
- Anti-EpCAM functionalized graphene oxide vector for tumor targeted siRNA delivery and cancer therapy
- Role of the cation-chloride-cotransporters in the circadian system