
TDP-43 and amyloid precursor protein processing: implications for Alzheimer’s disease
2021-12-09 14:12DavidHicks
中国神经再生研究(英文版) 2021年7期
David A. Hicks
In this perspective article, I will discuss our recent publication (Hicks et al., 2020),specifically its major findings and integration with the published literature. Alzheimer’s disease (AD) is a progressive neurodegenerative d i s e a s e o f p re d o m i n a nt l y u n k n ow n aetiology. Its neuropathological hallmarks are extracellular plaques formed of the amyloid beta (Aβ) peptide and neurofibrillary tangles of tau protein. Aβ peptides (Aβ1–40and Aβ1–42are considered the most important)are generated by sequential proteolysis of the amyloid precursor protein (APP). This proteolysis can take one of two pathways:the nonamyloidogenic or amyloidogenic(Figure 1). The former is mediated by α-secretase, predominantly a disintegrin and metalloprotease domain-containing 10(ADAM10) and the γ-secretase complex.This pathway releases the APP ectodomain as sAPPα, but crucially, results in cleavage of APP within the Aβ region, abrogating its generation. However, the amyloidogenic pathway is mediated by β-site APP cleaving enzyme 1 (BACE1, also known as β-secretase)and the γ-secretase complex, which results in the generation of Aβ species and the amyloid precursor protein intracellular domain. The amyloid precursor protein intracellular domain(AICD) has been shown to traffic to the nucleus where it can act as a transcriptional modifier.Although there are other minor APP secretases,those outlined herein represent the most significant. Alterations in APP processing have been linked to AD, insofar as the Aβ1–42: Aβ1–40ratio is increased in the disease (Andrew et al.,2016).
Over the last several years, transactive response DNA-binding protein 43 (TDP-43)has been linked to AD (Josephs et al., 2016).TDP-43 is a nucleic acid binding protein, which has a predominantly nuclear localization under physiological conditions (Budini and Buratti,2011). However, the formation of cytoplasmic inclusions of TDP-43 has been associated with several neurodegenerative disorders(Gao et al., 2018). These inclusions have been found in the brains of individuals with AD and their presence correlates inversely with cognitive performance (Josephs et al., 2016).In addition, a recent study has linked TDP-43 pathology in AD with the APOE gene. This study found a significant association between APOE genotype and TDP-43 proteinopathy stage.TDP-43 burden was found to be significantly correlated with the APOE ε4 allele (Yang et al., 2018). Although there is a multitude of neuropathological evidence linking TDP-43 to AD, mechanistic understanding is sparse. This refers to the direct effects exerted by TDP-43,which may initiate or potentiate AD pathology.These direct roles of TDP-43 in AD include dysfunction in mitochondria, RNA splicing,axonal transport, stress granules and protein quality control (Gao et al., 2018). This definition therefore excludes suggested roles of TDP-43 in AD, which may be secondary effects of, for example, neuroinflammation. Thus, our study sought to investigate putative links between TDP-43 and APP processing in the context of AD(Hicks et al., 2020).
Localization of TDP-43 and APP inneurons:Initially, we examined the possible co-localization of TDP-43 and APP to assess a possible direct interaction.Immunocytochemistry in human neurons(i.e. derived from induced pluripotent stem cells) confirmed a nuclear localization of TDP-43, but this was in complete opposition t o A I C D i m m u n o re a c t i v i t y. F u r t h e r immunocytochemistry and pharmacological treatment revealed AICD was localized to nucleoli, from which TDP-43 was excluded.Overall, this suggested that TDP-43 and APP do not interact directly.
Effects of APP expression on TDP-43:We subsequently assessed whether the APP holoprotein could modulate TDP-43 expression.This was performed by overexpressing each of the isoforms of APP (APP695, APP751and APP770)and analyzing the effects on TDP-43 expression.We also targeted APP using siRNA and found that neither over-expression nor knockdown of APP had a significant effect on TDP-43 expression.
Modulation of TDP-43 expression and effect on APP metabolism:TDP-43 is known to regulate its own expression via its 3’UTR(Budini and Buratti, 2011), presenting a challenge in generating an over-expression model. To address this, we generated a doxycycline-responsive inducible system,whereby TDP-43-FLAG was expressed upon doxycycline treatment. Using this system,TDP-43 was over-expressed and its ability to modulate APP processing was investigated.Initially, this involved immunoblot analysis of the APP holoprotein, ADAM10, BACE1 and the γ-secretase components (presenilin 1, presenilin 2, nicastrin and Pen2). Taken together, TDP-43 overexpression did not affect the immunoreactivity of any of these target proteins. This showed that TDP-43 does not modulate APP processing at the level of protein expression. Further work focused on the amyloidogenic processing pathway.Doxycycline-induced TDP-43 over-expression did not affect BACE1 mRNA levels or activity,nor did it alter levels of either Aβ1–40or Aβ1–42.
Role of TDP-43 in AD:Beyond a modest number of gene mutations, the aetiology of AD is unknown. Thus, novel avenues of investigation, such as TDP-43, are particularly important. Several years of research findings have reported on the deleterious effects of TDP-43 inclusions in AD, but there has been a notable absence of detailed mechanistic understanding. It has been suggested that TDP-43 may influence BACE1 expression(Herman et al., 2012), although this was in the context of increased TDP-43 induced neuroinflammation. This makes it difficult to delineate the effects of TDP-43 from those of generalized inflammation. Our work in cultured cells suggests that TDP-43 does not modulate either expression of APP or its processing(Hicks et al., 2020). Overall, this illuminates a general confounding feature of mouse models when compared to cultured human cells. Although the presence of non-neuronal cells in mouse models can be advantageous given their importance in brain function and dysfunction, these cells can obscure effects in neurons. Cell stress (such as over-expression of TDP-43) may activate microglia, resulting in downstream effects in neurons. In mouse models, it is consequently difficult to separate the direct effects of stress on neurons and the indirect effects mediated by non-neuronal cells.Our findings align with the finding that TDP-43 pathology is regionally disparate from the canonical amyloid pathology of AD (Josephs et al., 2016). Taken together, these data suggest that TDP-43 and Aβ may represent twin, nonoverlapping pathologies in AD. Were this to be the case, it would strengthen the growing case for multiple therapeutic interventions to target different pathological elements of the disease.For example, recent γ-secretase or Aβ targeting interventions have not proved successful in clinical trials. This may be because a substantial number of individuals with AD also have TDP-43 pathology, which is not addressed by amyloidfocused therapies.
Mitochondrial dysfunction:Recently, more information has started to emerge about the possible pathological effects of TDP-43 in AD.This has mainly, but not exclusively focused on putative deleterious effects of its dysfunction on mitochondria. Although TDP-43 has a predominantly nuclear localization, it is also associated with mitochondria, acting to regulate endoplasmic reticulum mitochondrial tethering as well as mitochondrial mRNA transport and protein translation. Furthermore, TDP-43 may be involved in mitochondrial fission/fusion and trafficking such that any disturbance in TDP-43 function could negatively impact on any of these key features of mitochondrial activity(Gao et al., 2018). Recently, an inhibitory peptide has been shown to be beneficial in an AD transgenic mouse model (Gao et al., 2020).This represents an early example of a possible TDP-43 centric therapy, which may be used in combination with more traditional amyloidfocused approaches.
Axonal transport:As mentioned, TDP-43 is associated with mitochondrial transport and this process along axons is disturbed in AD. However, dysfunction in TDP-43 axonal transport has also been associated with reduced trafficking of mRNA-containing TDP-43 granules. This may be linked to the role of TDP-43 in delivery of mRNAs for dendritic local translation (Gao et al., 2018). Taken together, these data suggest that reduced TDP-43 function in AD may lead to reduced mitochondrial and mRNA granule trafficking.
Stress granules:The characteristic TDP-43 inclusions seen in AD have also been associated with stress granules (SGs), although the precise relationship is unclear. SGs are formed in response to stress to repress translation of non-essential proteins. Some reports have suggested that SGs can potentiate TDP-43 aggregation, or that TDP-43 contributes to SG formation and slows their disassembly.However, the full role of TDP-43 in such stress response pathways remains to be elucidated(Gao et al., 2018).
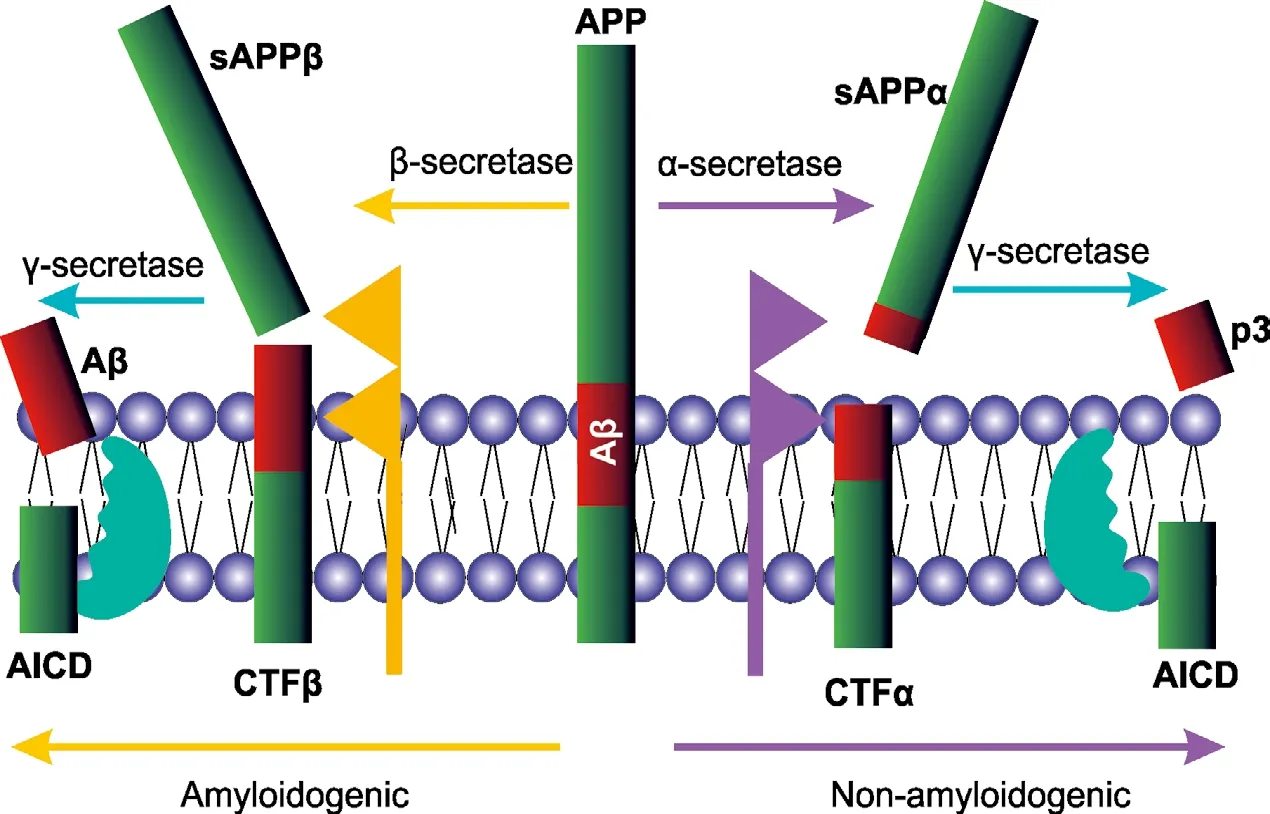
Figure 1|Processing of APP by α- and β-secretases.
Protein quality control:Recent data suggest that cytoplasmic TDP-43 aggregations may lead to impairment in the ubiquitin-proteasome system and this is dependent on protein tyrosine kinase 2. Protein tyrosine kinase 2 phosphorylates and activates TBK1, which then phosphorylates p62/SQSTM1, reducing the efficiency of protein aggregate transfer to the autophagosome (Lee et al., 2019). Interestingly,p62/SQSTM1 has been cited as a bridge between the ubiquitin-proteasome system and autophagy. A direct interaction of TDP-43 with p62/ SQSTM1 has been reported, which is disturbed in some cases of frontotemporal lobar degeneration (Gao et al., 2018).
RNA splicing:A general increase in cryptic exon incorporation has been demonstrated in AD brains and associated with TDP-43 dysfunction(Sun et al., 2017). A recent report showed dual phosphomimetic mutations in TDP-43 (T153E,Y155E) reduced splicing ability and binding affinity for A(UG)6RNA (Li et al., 2017). In the context of AD, TDP-43 has been shown to regulate the splicing of tau, promoting exon 10 inclusion and increasing the ratio of 4R-tau to 3R-tau, although this was not evident in a small AD patient cohort (Gao et al., 2018).
Conclusion:Thus, going forward, there is an urgent need to understand the precise role of TDP-43 in AD. For example, the presence of TDP-43 inclusions is a hallmark of motor neuron disease and, to a lesser extent,frontotemporal dementia. Given the low level of gene mutations driving TDP-43 pathology in motor neuron disease or AD (Ling et al.,2013), it is possible that similar processes lead to cytoplasmic TDP-43 accumulation in both diseases. However, this leads to the question of why such different neuronal populations are affected in the two diseases. In addition, AD can be present with or without TDP-43 pathology in relatively equivalent case numbers. This may be explained by a trigger which leads to Aβ and tau pathology. Then, in some individuals, a second“hit” could lead to aberrant accumulation of cytoplasmic TDP-43. This would go some way to explaining the worse cognitive performance of individuals with AD and TDP-43 pathology,compared to those without TDP-43 pathology.Indeed, a “two-hit” hypothesis has been discussed in the context of AD for some time.Several recent reports have revealed several putative roles for TDP-43 in AD, both in the context of loss of function and toxic gain of function, for example, through formation of cytoplasmic inclusions. Although TDP-43 has been linked to many cellular functions, this article is limited to those for which TDP-43 has a direct, AD-linked role. At the current time,mitochondrial function, axonal transport,protein quality control, stress granules and RNA splicing are key areas of investigation (Gao et al., 2018).
Overall, the data from our recent study(Hicks et al., 2020) show that,in vitro, TDP-43 does not modulate the expression of APP or its secretases (or components thereof).This suggests that the potential pathological effects of the cytoplasmic TDP-43 inclusions found in AD are distinct from those mediated by Aβ. Given the persistent pharmaceutical disappointments in this area, our findings suggest that failure to address TDP-43 pathology in AD may compromise the overall therapeutic strategy.
David A. Hicks*, †
Division of Neuroscience and Experimental Psychology, School of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester Academic Health Science Centre, Manchester, UK
†Current address: RBMOnline, Bourn Hall, Bourn,Cambridge, UK
*Correspondence to:David A. Hicks, PhD,david.hicks-2@manchester.ac.uk.
https://orcid.org/0000-0001-6045-1063(David A. Hicks)
Date of submission:May 14, 2020
Date of decision:June 21, 2020
Date of acceptance:September 7, 2020
Date of web publication:December 7, 2020
https://doi.org/10.4103/1673-5374.300983
How to cite this article:Hicks DA (2021) TDP-43 and amyloid precursor protein processing:implications for Alzheimer’s disease. Neural Regen Res 16(7):1402-1403.
Copyright license agreement:The Copyright License Agreement has been signed by the author before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Clusterin: a multifaceted protein in the brain
- Positive effects of music therapist’s selected auditory stimulation on the autonomic nervous system of patients with disorder of consciousness: a randomized controlled trial
- Transcranial pulse current stimulation improves the locomotor function in a rat model of stroke
- Comparative transcriptomic analysis of rat versus mouse cerebral cortex after traumatic brain injury
- Delayed atomoxetine or fluoxetine treatment coupled with limited voluntary running promotes motor recovery in mice after ischemic stroke
- Extremely low frequency electromagnetic fields promote cognitive function and hippocampal neurogenesis of rats with cerebral ischemia