
Dynamic glial response and crosstalk in demyelinationremyelination and neurodegeneration processes
2021-12-09 14:12TianciChuLisaShieldsWenxinZengYiPingZhangYuanyiWangGregoryBarnesChristopherShieldsJunCai
中国神经再生研究(英文版) 2021年7期
Tianci Chu, Lisa B.E. Shields, Wenxin Zeng, Yi Ping Zhang, Yuanyi Wang,Gregory N. Barnes, , Christopher B. Shields, , Jun Cai, ,
Abstract Multiple sclerosis is an autoimmune disease in which the immune system attacks the myelin sheath in the central nervous system. It is characterized by blood-brain barrier dysfunction throughout the course of multiple sclerosis, followed by the entry of immune cells and activation of local microglia and astrocytes. Glial cells (microglia,astrocytes, and oligodendrocyte lineage cells) are known as the important mediators of neuroinflammation, all of which play major roles in the pathogenesis of multiple sclerosis.Network communications between glial cells affect the activities of oligodendrocyte lineage cells and influence the demyelination-remyelination process. A finely balanced glial response may create a favorable lesion environment for efficient remyelination and neuroregeneration. This review focuses on glial response and neurodegeneration based on the findings from multiple sclerosis and major rodent demyelination models. In particular, glial interaction and molecular crosstalk are discussed to provide insights into the potential cell- and molecule-specific therapeutic targets to improve remyelination and neuroregeneration.
Key Words: astrocyte; crosstalk; demyelination; glial response; microglia/macrophage;multiple sclerosis; neurodegeneration; neuroinflammation; oligodendrocyte lineage cells;remyelination
Introduction
Multiple sclerosis (MS) is the most common chronic inflammatory and neurodegenerative disease affecting over 2.1 million individuals worldwide (Dilokthornsakul et al., 2016).The pathological hallmarks of MS include demyelination,gliosis, and neuroaxonal degeneration. They are caused by immune cell infiltration across the compromised blood-brain barrier (BBB) and inflammation in the central nervous system(CNS) (Dendrou et al., 2015). The loss of homeostasis and the activation of glial cells play essential roles in the progression of demyelination and neuronal/axonal injury. The interactions between glial cells, especially the effects on oligodendrocyte progenitor cells and oligodendrocytes (OPCs/OLs), affect the demyelination-remyelination process by a cascade of complex responses and signalings (Miron, 2017; Zrzavy et al., 2017;Baaklini et al., 2019).
The major glial cell population in the adult vertebrate CNS includes OPCs/OLs, microglia, and astrocytes. There are approximately 60 billion glial cells in the human brain based on a validated quantification with isotropic fractionator (von Bartheld et al., 2016). They orchestrate CNS homeostasis and support transmission of chemical and electrical signals via network communication under physiological and demyelinating conditions (Clemente et al., 2013; Domingues et al., 2016; Baaklini et al., 2019). The spatio-temporal patterns of glial activation and phenotype are dynamic in the pathogenesis of demyelination (Miron et al., 2013;Chu et al., 2019). Complex glial interactions and molecular conversation regulate the behaviors of OPC/OLs and affect the demyelination-remyelination process (Clemente et al., 2013;Domingues et al., 2016; Miron, 2017; Baaklini et al., 2019; Jha et al., 2019).
Studies cited in this review were searched on the PubMed and Google Scholar databases between January and March in 2020, using mainly the search terms: astrocyte, microglia,macrophage, oligodendrocyte, glial response, molecular interaction, multiple sclerosis, demyelination model,neurodegeneration, remyelination, and various possible combinations of those terms.
This review summarizes the current knowledge in the glial response and neurodegenerative changes during the pathogenesis of MS and major rodent demyelination models.Cellular and molecular crosstalks are highlighted to reveal how glial cells are orchestrated and potential intervention targets during the processes of demyelination and neurodegeneration.
Glial Features in Demyelination-Remyelination Process and Neurodegeneration
OLs occupy approximately 45–75% of the total glial cell population in the human brain (von Bartheld et al., 2016).They are the vital myelin-forming cells that allow saltatory conduction along axons and provide trophic support for axons(Dulamea, 2017). Loss of OLs and/or myelin sheaths are the predominant pathological features of demyelinating diseases such as MS resulting in loss of neural functions (Barnett and Prineas, 2004). OPCs are widely distributed throughout the adult CNS and, due to their capacity to proliferate, migrate,and differentiate into mature OLs, serve as an endogenous resource for remyelination (Bø et al., 2013; Crawford et al., 2013). Remyelination occurs in MS, however, its limited capacity contributes to progressive neurodegeneration and irreversible clinical impairment (Boyd et al., 2013; Campbell and Mahad, 2018). Remyelination may fail due to inadequate OPC recruitment and arrested OPC differentiation, which are related to a variety of factors such as age, disease duration,myelin debris clearance, and the balance between proregenerative and inhibitory factors in MS lesions (Boyd et al.,2013; Frischer et al., 2015; Peferoen et al., 2016; Cunniffe and Coles, 2019).
Microglia constitute 10% or less of glial cells in the human brain (von Bartheld et al., 2016). Macrophages in the CNS arise from resident microglia and blood-derived monocytes.Macrophages predominate in demyelinating lesions in MS and MS models, and their accumulation correlates to disease severity along with activated microglia (Trapp et al.,1998; Bogie et al., 2014; Yamasaki et al., 2014; Zrzavy et al.,2017). Though microglia-derived and monocyte-derived macrophages are ontogenetically different and may function distinctly, they are indistinguishable by light microscopy and surface phenotypes (Yamasaki et al., 2014). Additionally,activated microglia and macrophages share many functional similarities in orchestrating neuroinflammation including antigen presentation, phagocytosis of myelin debris and apoptotic cells, as well as production of a broad range of factors including cytokines, chemokines, reactive oxygen species (ROS), and secondary messengers (DiSabato et al.,2016; O’Loughlin et al., 2018; Baaklini et al., 2019). The temporal and spatial patterns of activated microglia and macrophages are closely related to the progression and modulation of demyelination and neurodegeneration (Miron et al., 2013; O’Loughlin et al., 2018; Chu et al., 2019). Due to the resemblance in phenotype and function in diseased status, microglia and macrophage are referred to collectively as M/M in this review.
Astrocytes comprise 19–40% of the total glial cells in the human brain (von Bartheld et al., 2016). They undergo dramatic transformation into reactive astrocytes in response to the demyelinating insult and become neurotoxic (Chu et al., 2017; Liddelow et al., 2017; Jha et al., 2019). They are also a major source of several molecules, including chemokines,growth factors, and pro- and anti-inflammatory cytokines.Reactive astrocytes play key but controversial roles in driving inflammation and forming astrocytic gliosis which exert both detrimental and beneficial effects during progression and recovery of demyelination and neurodegeneration (Skripuletz et al., 2013; Brambilla et al., 2014).
Glial Response and Plaques in Multiple Sclerosis Spatial pattern in MS plaques
The pathological hallmarks of MS include multiple sites of focal demyelinating plaques in the CNS combined with varying degrees of inflammation and gliosis, oligodendrocyte loss, and diffuse neurodegeneration (Popescu et al., 2013; Lassmann,2018). Demyelinating plaques in MS are classified according to their pathohistological features based on glial density and spatial pattern: 1) active (acute), 2) chronic-active, and 3) chronic-inactive (Table 1). Lesions with greater than 60%remyelination are defined as shadow plaques (Patrikios et al.,2006).
The active plaque is most frequently found in acute and relapsing-remitting MS (RRMS) (Popescu et al., 2013).It is a hypercellular demyelinating lesion with indistinct margins and massive myelin-laden M/M throughout the lesion, indicating myelin degradation activity in the plaque.Astrocytes proliferate and become hypertrophic with abundant cytoplasm and an increased number of fibrillary processes but without significant astroglial scar (Frohman et al., 2006; Popescu et al., 2013). Loss of OLs is observed in the active plaque accompanied by a sufficient number of OPC (Frohman et al., 2006; Boyd et al., 2013). Widespread axonal degeneration occurs, recognized by the presence of terminal axonal swelling, ovoids of amyloid precursor protein (APP) accumulation in axon terminals, and many axons labeled with anti-non-phosphorylated neurofilament antibody (SMI32 positive; Trapp et al., 1998, 1999). APP accumulation is recognized as an early and sensitive marker of acute axonal dysfunction, and its magnitude correlates with the degree of inflammation (Wilkins and Scolding, 2008).Neurofilament is a major structural component of axons, and its phosphorylation contributes to axonal structure, alignment,transport, and stability. Accumulation of APP represents a functional change in axonal transport, however, neurofilament dephosphorylation reflects a structural change in axons and predisposes them to degeneration (Wilkins and Scolding,2008).
Chronic plaques are frequently observed in progressive MS. The chronic-active plaque is sharply demarcated with a hypocellular demyelinated core surrounded by an active demyelinating edge with activated M/M (Trapp et al., 1999;Kornek et al., 2000; Frohman et al., 2006; Bramow et al.,2010). Astrogliosis is present in the center with hypertrophic astrocytes distributed at the margins (Frohman et al., 2006).Most chronic-active plaques show loss of both OLs and OPCs, and the density of OPCs varies in different parts of the lesion. OPC insufficiency is observed in approximately 37%of the chronic-active MS plaques (Boyd et al., 2013). This finding indicates that insufficient OPC recruitment may be an important reason for the remyelination failure in chronicactive plaques (Lucchinetti et al., 1999; Frohman et al., 2006;Boyd et al., 2013). Axonal APP accumulation and the number of SMI32 positive spheroids are low in the center but higher at the slowly expanding border of chronic-active plaques (Trapp et al., 1999; Kornek et al., 2000; Frohman et al., 2006).
Chronic-inactive plaques are a completely demyelinated hypocellular lesion with a sharp border. There are few M/M cells, but profound astrogliosis exists within the lesion. Though most chronic-inactive plaques contain a sufficient number of OPCs, there is a substantial loss of oligodendrocytes and axons, suggesting that arrested maturation may be the main cause of remyelination failure in chronic-inactive plaques(Lucchinetti et al., 1999; Frohman et al., 2006; Boyd et al.,2013; Popescu et al., 2013). There is minimal APP and SMI32 staining, implying a low level of ongoing axonal damage within the chronic-inactive plaque (Kornek et al., 2000; Wilkins and Scolding, 2008).
Remyelinated plaques are sharply demarcated and characterized by the reappearance of OPC/OL and uniformly thin myelinated axons. These plaques are defined as shadow plaques if remyelination has occurred in over 60% of the total lesion area (Lucchinetti et al., 1999; Kornek et al., 2000;Patrikios et al., 2006; Bramow et al., 2010). Few M/M and a minor amount of astrogliosis are identified in remyelinated plaques (Lucchinetti et al., 1999; Kornek et al., 2000; Patrikios et al., 2006; Bramow et al., 2010). There is a moderate reduction in axonal density, but no significant axonal damage exists throughout the remyelinated shadow (Kornek et al.,2000). Although remyelination may protect axonal integrity and restore saltatory conduction (Kornek et al., 2000),remyelinated plaques are more vulnerable to recurrent attacks of demyelination than normal-appearing white matter (NAWM)(Bramow et al., 2010). Shadow plaques seem to occur most frequently in patients with RRMS and primary progressive MS(PPMS) compared to those with secondary progressive MS,though there is no statistically significant difference between these findings (Patrikios et al., 2006). The patients with a longer disease duration who died at an older age are significantly more frequently associated with more remyelinated lesion areas, suggesting that individual variability and extent of remyelination should be considered in developing therapeutic strategies for MS (Patrikios et al., 2006).
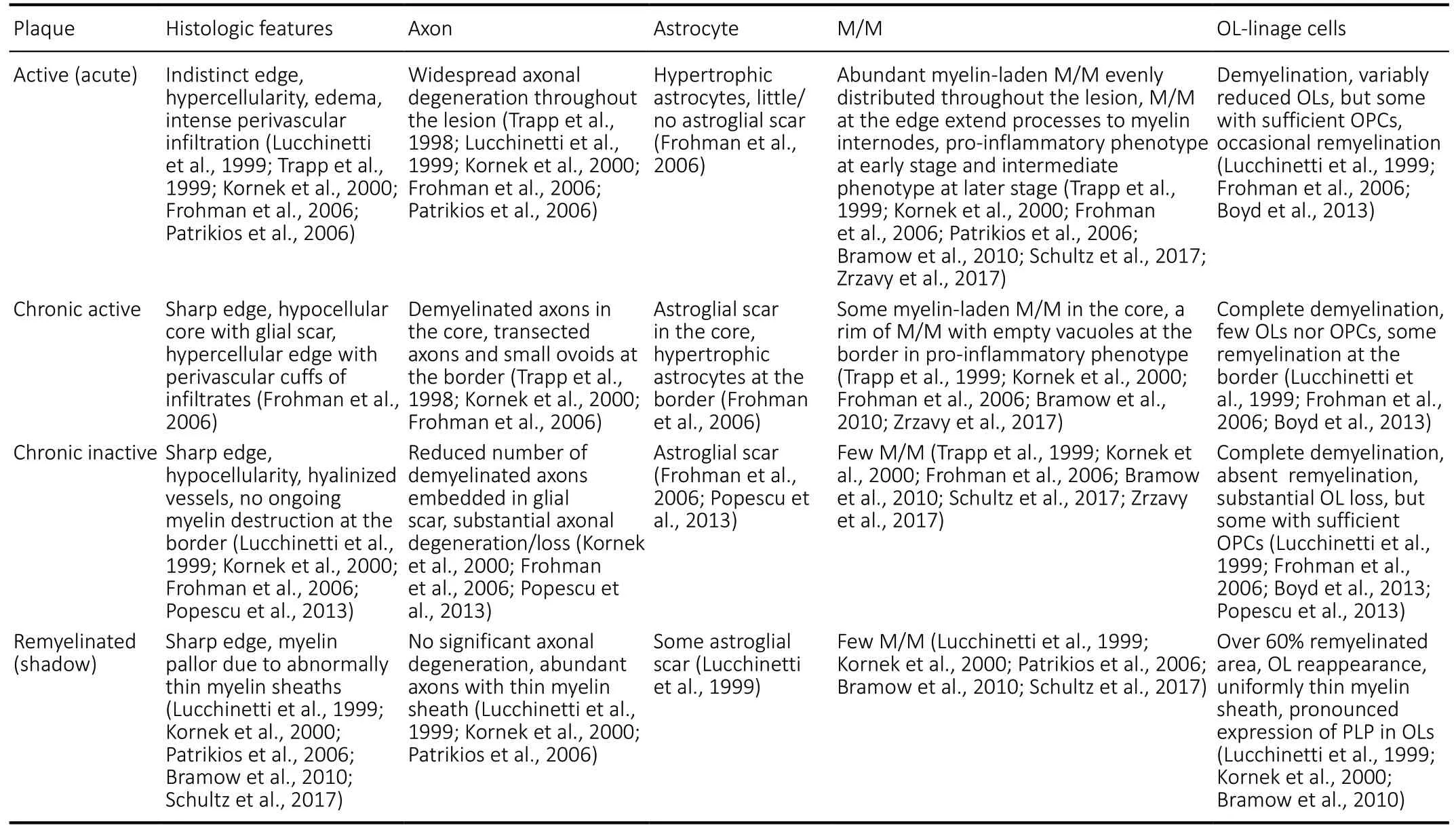
Table 1 |Glial response and neurodegeneration in multiple sclerosis plaques
Glial response in MS plaques
Pathological studies suggest that inflammation drives initial demyelination and neurodegeneration in all stages of MS.This damage is further exacerbated by a secondary wave of inflammation, mediated by various chemokines and adhesion molecules arising from infiltrates and activated glial cells in response to the initial injury (Lassmann, 2014, 2018). Thus,anti-inflammatory strategy was considered a favorable therapy to promote remyelination and protect axons in MS. However,current treatment by global immunosuppression may be inefficient in preventing neurodegeneration and decreasing disease progression (Smith and Cohen, 2016; Lorscheider et al., 2019). As major effectors of neuroinflammation, M/M and astrocytes play complex roles in the progression and resolution of demyelination and neurodegeneration (DiSabato et al.,2016; Miron, 2017; Zrzavy et al., 2017; O’Loughlin et al., 2018).
M/M are significantly increased and present a proinflammtory phenotype (with the gene expression profile of markers (CD68, p22phox, MHC antigens, CD86) in early active lesions, expressing molecules involved in phagocytosis,antigen presentation, oxygen radical production, and T cell co-stimulation (Zrzavy et al., 2017). These M/M are declined and altered to an intermediate activation status at later stages during lesion maturation. A similar increase and activation in M/M are found restricted at the active demyelinating rim in chronic and slowly expanding MS lesions, while few M/M are present in the inactive lesion core or inactive lesion (Frischer et al., 2009; Zrzavy et al., 2017). Although M/M-mediated inflammation and oxidative stress induce demyelination and axonal injury in MS lesions, there exists a positive relationship between inflammation and remyelination in MS tissues(Lassmann et al., 2012). M/M may exert pro-regenerative effects by producing anti-inflammatory and neuroprotective factors and phagocyting myelin debris from injury sites,which facilitate the recruitment and differentiation of OPCs necessary for efficient remyelination (Kotter et al., 2005; Bogie et al., 2014; Lampron et al., 2015). This evidence indicates that M/M play a multifaceted and phenotype/functionalstatus dependent role in the pathogenesis of MS, and the eventual outcome is related to a re-balance between the secreted immunomodulatory mediators.
Astrocyte activation is evident starting from the initial stage of MS and is a highly active contributor to lesion formation(Ludwin et al., 2016; Ponath et al., 2018). The major roles of activated astrocytes in MS include: secreting and responding to a host of neurotoxic/pro-inflammatory and neuroprotective/anti-inflammatory mediators (cytokines,chemokines, and growth factors); regulating oxidative stress by not only producing ROS and reactive nitrogen species but also neutralizing them with antioxidant enzymes like superoxide dismutases and NAD(P)H:quinone oxidoreductase 1 (NQO1);impairing BBB integrity with disrupted end-feet connection to vasculature and increasing BBB permeability with astrocytic expression of angiogenic vascular endothelial growth factor A; and forming a glial scar after acute inflammation in chronic active and chronic inactive lesions, which may be a barrier to neuroregeneration but also a beneficial structure to prevent the spread of infiltrates and neurotoxic factors,as well as to support demyelinated axons and restore BBB function (Brosnan and Raine, 2013; Brambilla, 2019).Although a positive correlation exists between astrocyte activation and disease extent and severity (Brosnan and Raine, 2013), ablation or disruption of reactive astrocytes did not prevent myelin/axonal damage but increased infiltration and inflammation, delayed M/M recruitment, and reduced removal of myelin debris, resulting in severe demyelination and neurodegeneration (Voskuhl et al., 2009; Skripuletz et al.,2013). To improve remyelination in the context of MS that is hostile to oligodendrocyte lineage cells especially OPCs (Cui et al., 2013; Kirby et al., 2019), it is important to identify/regulate interacting factors secreted by M/M and astrocytes to guide OPC/OL behaviors in an appropriate sequence of activation, recruitment, and differentation into myelin sheath-forming OLs. Glial responses during MS pathogenesis are dynamic, and their effects are multifaceted. Instead of global immunosuppression, selective modulation based on the knowledge of glial response patterns (temporal and spatial) during the development of MS may be a preferred strategy for efficient remyelination and neuroprotection. This strategy may promote regeneration by re-balancing the lesion context with increased pro-regenerative factors and reduced inhibitory factors at the right timing to improve the milieu and to positively regulate OPC/OL behaviors.
Neuro-axonal damage and loss contribute to disease progression and persistent clinical impairment in patients with MS (Singh et al., 2017). Axonal degeneration develops through multiple mechanisms including anterograde (Wallerian) and retrograde (dying back) degeneration (Simons et al., 2014).Focal axonal damage caused by loss of trophic support from oligodendrocytes/myelin and disruption in axonal transport result in axonal transection and Wallerian degeneration in MS(Kornek et al., 2000; Nikic et al., 2011). Axonal degeneration in active demyelination occurs more frequently than in other plaque types (average number of terminal axon ovoids per cubic millimeter: 11,236 in active lesions, 3138 at the border of active lesions, 875 at the center of chronic-active lesions, and less than 1 in normal white matter) (Trapp et al., 1998). Axonal degeneration correlates with the severity of inflammation in early MS (Trapp et al., 1998; Kornek et al., 2000). Axonal degeneration and loss may proceed in the absence of acute inflammation in chronically demyelinated axons. This results in progressive axonal loss and clinical deterioration in patients with chronic progressive MS who show no evidence of relapse on MRI scans, cerebrospinal fluid pleocytosis, or response to anti-inflammatory treatment(Trapp et al., 1998, 1999). Additionally, in a cohort study of patients with MS in early and late stage of remyelination, no statistical difference was found in the density of preserved and damaged axons between demyelinating and remyelinating plaques in early stages of lesion formation. However, the number of preserved axons was significantly higher in remyelinated plaques than demyelinated plaques at a late stage. This finding suggests that remyelination is protective and essential to the recovery of acute axonal damage after a demyelinating insult (Schultz et al., 2017). More importantly,improved remyelination correlates with less clinical disability in patients with MS (Bramow et al., 2010; Bodini et al., 2016).This evidence suggests that axonal damage in early MS is a result of primary demyelination. Pro-remyelination strategies appear to be reasonable therapeutic approaches and are showing benefit in recent clinical trials (Plemel et al., 2017).
Glial Response in the Demyelination-Remyelination Process and Neurodegeneration:Evidence from Experimental Demyelination Models
Most advances in understanding MS pathogenesis arise from experimental animal models. Well-characterized experimental models of demyelination and remyelination include experimental autoimmune encephalomyelitis (EAE), viralinduced chronic demyelinating disease like Theiler’s murine encephalomyelitis virus (TMEV) infection, toxin-induced systemic demyelination (Cuprizone), and toxin-induced focal demyelination such as L-α-lysophosphatidylcholine (LPC) and lipopolysaccharide (LPS). Although a perfect animal model does not exist since MS is a human disease with complex and heterogeneous pathological features, the use of different animal models allows us to understand distinct aspects of MS instead of its entire complexity and to investigate possible mechanisms and potential treatments. Among them, EAE is considered most clinically relevant because of its similarities to MS in histopathology and immunology. Lymphocyte-mediated demyelination and axonal degeneration with clinical signs can be induced by immunization with peptide antigens like myelin oligodendrocyte glycoprotein or spinal cord homogenade in susceptible strains to mimic the chronic disease course of MS (Lassmann and Bradl, 2017). Chronic EAE in Biozzi ABH mice is particularly relevant to MS with a relapsing-remitting episode followed by a secondary progressive disability. This model effectively studies disease progression and evaluates therapeutic strategies targeting this process (Hampton et al., 2008; Jackson et al., 2009; Al-Izki et al., 2012). However,EAE model may not be appropriate to study remyelination considering that the ongoing and recurrent inflammation and demyelianation may confound the evaluation of remyelination. Unlike MS and EAE model, systemic or focal toxin-based models induced by cuprizone, LPC, or LPS cause little lymphocytic response nor clinical signs. Yet, they create lesions with precise timing and spatial patterns, which provide a simpler system than immune-mediated models to study cellular response and regenerative biology as well as screen potential therapeutic targets during the demyelinationremyelination process (McMurran et al., 2019). These models cover a broad spectrum of MS phenotypes and provide an understanding of the glial response in demyelination,remyelination, and neurodegeneration at both the cellular and molecular level. The dynamic characteristics of glial activation and response in major rodent models are summarized inTable 2. The differences between models and their responses are important knowledge for the choice of appropriate models and the interpretion of results. A combination use of models based on their advatanegs and weakness may benefit MS studies.
M/M response in the demyelination-remyelination process and neurodegeneration
Similar to active MS plaques, M/M are the most abundant immune cells in demyelinating lesions. M/M adopt a homeostatic phenotype in the resting state and are highly responsive sensors to damages, playing a surveillance role in the CNS (Gonzalez et al., 2014; O’Loughlin et al., 2018).M/M present a different activation status depending on the stage of the disease. M/M were originally divided into classically activated/pro-inflammatory (M1) and alternatively activated/anti-inflammatory (M2) status: M1 are associated with host defense, phagocytosis, and antigen-presentation,which may be cytotoxic to OLs and exacerbate demyelination and neurodegeneration by producing pro-inflammatory mediators (tumor necrosis factor-α [TNF-α], interleukin [IL]-1β,interferon-γ (IFN-γ), ROS, RNS, and complement proteins) and chemokines of the CC and CXC families (Gonzalez et al., 2014;Zrzavy et al., 2017; O’Loughlin et al., 2018). M2 are associated with remyelination and neuroprotection by secreting antiinflammatory cytokines (IL-4, IL-13, IL-10, transforming growth factor-β) and neurotrophic factors (nerve growth factor, brainderived neurotrophic factor, and insulin-like growth factor 1)(Miron et al., 2013; Gonzalez et al., 2014; O’Loughlin et al.,2018). With the advance in omic technologies, this dichotomy is simplified as new evidence indicates that the transcriptional changes in M/M are varied and context-dependent (Ransohoff,2016; van der Poel et al., 2019). M/M have heterogenous subsets and undergo disease-specific transformations during aging and in models of MS and other neurodegenerative diseases (Mrdjen et al., 2018). It is important to take variables like age, disease stage, and brain region into consideration to define the phenotypic changes in M/M. Although it is an oversimplified classification, the M1/M2 polarization is a starting point to study the roles of M/M activation during the demyelination-remyelination process. Markers used in studies to identify M/M phenotypes are detailed for reference considering that M1/M2 only depict two polars of the inflammatory responses.Polarization follows myelin phagocytosis and is predominantly observed in the inactive lesion center (Miron et al., 2013;Zrzavy et al., 2017). The M/M phenotypic switch influences demyelination severity and remyelination efficacy, presumably by alternating the cytokine expression and OPC response(Kotter et al., 2005; Olah et al., 2012; Miron et al., 2013;Miron, 2017; O’Loughlin et al., 2018). Activation of the initial pro-inflammatory M1 is accompanied by OPC proliferation phase, and selective depletion of M1 reduces the density of proliferating OPC in the LPC-induced demyelination lesion(Miron et al., 2013). A shift from M1- (iNOS+/CD68+) to M2-(Arginase 1+/CD68+, CD206+/CD68+) dominant response occurs at the initiation of remyelination and drives OPC differentiation. Instead of direct phenotypic transition,necroptosis of pro-inflammatory M/M and repopulation of pro-regenerative M/M are required for the M/M shift and contribute to efficient remyelination in a cuprizone-induced model (Lloyd et al., 2019). Moreover, a unique phenotype localization was identified in a LPC-induced demyelination lesion for initial remyelination (Chu et al., 2019). The M2 phenotype (CD206+/Iba1+, Arginase 1+/Iba1+) is concentrated within the lesion core surrounded by OPC/OL, astrocytes,and the M1 phenotype (CD16/32+/Iba1+, CD86+/Iba1+). This spatial arrangement suggests that, besides the polarization phenotype, the regional change of M1 (outside; surveillance,barrier, pro-inflammation) and M2 (inside; regeneration,protection, anti-inflammation) may be another important factor for OPC differentiation and initial remyelination.

Table 2 | Glial response and neurodegeneration in rodent demyelination models
Astrocytic response in the demyelination-remyelination process and neurodegeneration
The appearance of a hypertrophic astrocyte at the plaque margin in acute active MS is one of the earliest pathological events (Brosnan and Raine, 2013). Although astrocytes are activated upon stimulation in most demyelination models(Table 2), their temporal and spatial patterns vary between models. Astrocytes are activated and proliferate throughout the LPS-induced demyelination lesion in the early and middle stages but is not observed in late stages (Felts et al., 2005;Chu et al., 2019). However, acute astrocytic loss is observed in early stages of LPC-induced demyelination followed by elevated activation and proliferation around the lesion core until remyelination occurs (Plemel et al., 2018; Chu et al.,2019). Importantly, reactive astrocytes produce a broad spectrum of cytokines and chemokines that modulate the lesion environment and undergo astrogliosis to form a glial scar in chronic MS plaques (Brosnan and Raine, 2013). A glial scar is customarily considered to inhibit remyelination and axonal regeneration. A scar provides transient neuroprotection by restricting infiltration and inflammatory processes, however, over the long-term it creates a physical and biochemical barrier including inhibitory molecules and proteoglycans (chondroitin sulfate proteoglycans, CSPGs) that impede OPC access, myelin repair, and neuronal outgrowth based on findings inin vitroand multiplein vivomodels including EAE, TMEV, and LPC models (Voskuhl et al., 2009;Keough et al., 2016; Feliu et al., 2017). However, therapeutic approaches to restrict astrocytic scar formation or removal of a chronic scar results in reduced axonal regrowth and impaired functional recovery after crush spinal cord injuries, indicating that astrogliosis aids and inhibits neuroregeneration (Anderson et al., 2016). Recent studies report that a glial scar does not prohibit remyelination in a rodent EAE model by immunizing Dark Agouti rats with myelin oligodendrocyte glycoprotein.Patterns of astrocyte-derived factors and the phenotype switch of reactive astrocytes from A1 (neurotoxic astrocytes)to A2 (ischemic astrocytes) during lesion development indicate that finely regulated and balanced astrogliosis may benefit remyelination (Haindl et al., 2019). In a LPC-induced demyelination model, dense astrogliosis surrounds the lesion core in a manner reminiscent of glial scar followed by robust remyelination after three weeks (Chu et al., 2019). These studies support a novel hypothesis that abundant astrogliosis after demyelination does not prohibit OPC survival, migration,or remyelination. Instead, it may provide a complex lesion environment that influences the response of OPCs/OLs and other glial and/or inflammatory cells rather than only creating a rigid inhibitory physical barrier. Astrocytic-derived molecules may play an essential (beneficial or adverse) role in modifying the extracellular microenvironment and regulating the demyelination-remyelination process.
Hypertrophic astrocytes secrete chemokines such as CCL2 and CXCL10 which activate both microglia and astrocytes in an autocrine or paracrine manner at the rim of chronicactive plaques in secondary progressive MS (Tanuma et al., 2006). Inhibition of astroglial nuclear factor-kappa B activation reduces peripheral immune cell infiltration and the number of total and activated M/M, contributing to alleviated disease severity, enhanced remyelination, and improved functional outcome following EAE. This may be related to a reduction in multiple proinflammatory cytokines(TNF-α, IFN-γ), chemokines (CXCL9, CXCL10, CCL2, CCL5),and cell adhesion molecules (ICAM-1, VCAM-1, integrin-β5,and integrin-β7), all of which contribute to the initiation and maintenance of the inflammatory response in EAE(Brambilla et al., 2009, 2014). Using the TMEV model that simulates progressive MS, it was observed that suppressing CSPG deposition in demyelinating lesions augments the number of mature OLs and increases myelin basic protein,which is beneficial to remyelination and motor functional improvement (Feliu et al., 2015, 2017). Hypertrophic astrocytes express high levels of chemoattractant molecules for OPC including CXCL8, CXCL1 and CXCL10 at the edge of active MS plaques, suggesting their important roles in promoting OPC recruitment and remyelination (Omari et al.,2005). Furthermore, the expression level of CXCL12, a vital chemokine that regulates OPC migration and differentiation, is significantly increased within reactive astrocytes in plaques of active MS. Upregulation of CXCL12 is also observed in reactive astrocytes and some M/M at the plaque edge in silent and chronic MS plaques, although its expression is lower than that in active plaques (Calderon et al., 2006; Moll et al., 2009;Chu et al., 2017). OL-mediated remyelination occurs in lesion areas where astrocytes are present, which contributes to the restoration of the CNS glial microenvironment. Schwann cellmediated remyelination occurs in regions where astrocytes are absent, which mediates tissue patches resembling the peripheral nervous system. This process signifies the crucial role of reactive astrocytes in the OPC/OL response and predicts the remyelination type that will occur (Monteiro de Castro et al., 2015). Rather than the entire inhibitory function of astrogliosis, alternative treatments that specifically target the beneficial or inhibitory molecules produced by reactive astrocytes may be novel and effective strategies to promote remyelination and neuroregeneration.
OPC/OL response in the demyelination-remyelination process and neurodegeneration
OPC are widely distributed throughout the adult CNS(Crawford et al., 2013). They react to myelin damage via proliferation, migration, and differentiation into myelinating OLs to enhance axonal function (Bø et al., 2013; Crawford et al., 2013; Chu et al., 2019). OPC are responsive to a broad range of chemokines, cytokines, mitogens, and growth factors that are released by activated astrocytes and M/M following demyelination (Clemente et al., 2013; Domingues et al., 2016; Baaklini et al., 2019). Among all the factors that may lead to remyelination failure, OPC/OL behavior such as inadequate OPC recruitment and arrested OPC differentiation during the pathogenesis of MS must be resolved to ensure efficient recovery (Boyd et al., 2013; Cunniffe and Coles,2019). Therefore, glial interactions and extracellular factors that affect OPC recruitment and differentiation may be essential to the extent of remyelination and recovery from neurodegeneration. Adult OPC transits into a reactive status and is reprogrammed into a more neonatal-like OPC,producing chemokine CCL2 and cytokine IL-1β that promote OPC mobilization and repopulation in demyelinated areas in cuprizone-treated mice (Moyon et al., 2015). A total of 3–5% of OPC express CCL2 within active MS plaques, whereas only 1-2% are detected in chronic MS plaques and NAWM.Moreover, a 2.4- and 1.7-fold increase of CCL2-expressing OPC were found within active MS plaques and in the active rim of chronic plaques, respectively (Moyon et al., 2015).
OLs release many other factors that improve neuronal survival and provide axon support, such as brain-derived neurotrophic factor, glial cell-derived neurotrophic factor(GDNF), and neurotrophin-3 (NT-3) (Dai et al., 2003; Wilkins et al., 2003). In response to MS and EAE, OPCs exhibit capacity to phagocytize myelin debris, and OL lineage cells(Olig2+, Olig1+, Sox10+) express several immunomodulatory factors associated with immune cells, including cytokines,chemokines, antigen presenting molecules, complement,complement regulatory molecules, and extracellular matrix proteins (Zeis et al., 2016; Falcao et al., 2018). OPCs highly express major histocompatibility complex (MHC) class I and present antigens to cytotoxic T cells when exposed to interferon-γ (IFN-γ), resulting in OPC death bothin vitroandin vivo(Kirby et al., 2019). These findings suggest that OPCs/OLs may actively contribute to the pathogenesis of MS by adjusting self-behaviors and affecting the immune response rather than being passively targeted. An understanding of the evolution of OPCs/OLs may stimulate development of innovative therapeutic strategies for demyelinating and neurodegenerative diseases.
Glial interaction and molecular crosstalk following demyelination
In response to pathological stimuli, bidirectional communication exists between activated M/M and reactive astrocytes which is mediated by diverse molecular secretions.M/M, usually responding faster than astrocytes, activate A1 by releasing IL-1α, TNF-α, and complement component 1qin vitroandin vivo(Liddelow et al., 2017). In MS, A1s are found at high levels in active plaques, although they remain present in active and inactive chronic plaques. A1 are closely associated with activated M/M in these plaques. A1 lose most normal astrocyte functions such as supporting neuronal survival and outgrowth support, phagocytosis, and synaptogenesisin vitro. Mature OLs and neurons do not survive in this altered astrocytic milieuin vitro. M/M are insufficient to kill neurons, however, they may induce A1 which may drive neurodegeneration and failure of synaptic formation by secreting neurotoxins and complement components (Liddelow et al., 2017). Alternatively, astrocytes regulate the recruitment of M/M and peripheral immune cells by secreting a spectrum of cytokines and chemokines. Reactive astrocytes secrete CCL2 and CXCL10 to recruit and active M/M, which benefits remyelination with improved myelin debris clearance in MS and cuprizone-induced demyelinating lesions (Tanuma et al.,2006; Skripuletz et al., 2013). Astrocytes also attenuate M/M activation and suppress inflammation by secreting factors like Orosomucoid-2 and GDNFin vitro(Rocha et al., 2012; Jo et al.,2017).
M/M- and astrocyte-derived molecules may affect OPC/OL behavior and affect the process of demyelination and neurodegeneration. Reactive astrocytes and M/M release guidance molecules like Semaphorin (Sema) 3A(chemorepellent) and 3F (chemoattractant) to control OPC migration within MS plaques (Boyd et al., 2013). Sema 3A and 3F are differently expressed in MS plaques. A higher expression of Sema 3F compared to 3A in active plaques with sufficient OPC contributes to a greater degree of inflammation with an increased opportunity for remyelination. Both reactive astrocytes and some M/M significantly upregulate the expression of CXCL12 in MS plaques which may benefit myelin repair by enhancing OPC recruitment and differentiation via the CXCL12/CXCR4/CXCR7 signaling axis (Calderon et al., 2006; Moll et al., 2009; Chu et al., 2017). The timing of releasing different molecules is important in regulating OPC behavior for remyelination. A switch from M1 (iNOS+/CD68+) to M2 (Arginase 1+/CD68+, CD206+/CD68+) polarization at 10 days post-injection in a LPC-induced demyelinating lesion corresponds to the onset of OPC differentiation. M2-derived activin-A (TGF-β superfamily member) and insulin-like growth factor-1 drive OPC differentiation at the appropriate time that contributes to efficient remyelination (Mason et al., 2001; Voss et al., 2012; Miron et al., 2013). Astrocytederived factors play a complex dual role in OPC response following demyelination. Reactive astrocytes overexpress bone morphogenetic proteins 2/4 that may block OPC differentiation via the bone morphogenetic protein signaling pathway, as well as endothelin-1 that may inhibit OPC differentiation by promoting Notch activation in OPCs during remyelination (See and Grinspan, 2009; Hammond et al.,2014). Reactive astrocytes also secrete ciliary neurotrophic factor, which promotes OPC proliferation via fibroblast growth factor 2 (FGF2) signaling and enhances OL maturation through the glycoprotein130-Janus kinase pathwayin vitroand in viral-induced demyelination (Stankoff et al., 2002;Albrecht et al., 2003). Of note, FGF2 is rapidly up-regulated within MS plaques (active plaque and the rim of the chronic active plaque) and NAWM by astrocytes and M/M (Clemente et al., 2011; Thummler et al., 2019). FGF2 by astrocytes is positively correlated with the severity of inflammation in MS plaques. It shows diametrically opposing functions in inhibiting myelination/remyelination but promotes OPC generation and proliferationin vitroandin vivo, which may be dependent on the stage-specific expression of FGF receptors 1–3 and their level of usage (Azim et al., 2012; Thummler et al., 2019). Therefore, any attempts to regulate FGF2 signaling with functional pleiotropy may promote its beneficial while suppressing its detrimental effects on remyelination. As a major extracellular matrix (ECM) modulator, astrocytes produce a variety of ECM-related molecules to affect OPC activities following demyelination. The accumulation of high molecular weight hyaluronan (Back et al., 2005), fibronectin(Stoffels et al., 2013), Tenascin-C (Czopka et al., 2010),and CSPGs (Keough et al., 2016) impairs remyelination by preventing OPC maturation and process outgrowth. The complex molecular interplay between glial cells not only suggests their multifaceted roles in demyelinating lesions but also provides potential cell-specific therapeutic targets during the demyelination-remyelination process.
It is important to note that many cellular responses and the roles of molecules mentioned above are dynamic and may vary with disease course, lesion type and severity, CNS region,oligodendrocyte lineage cell stage, and demyelinating lesion environment, all of which may result in controversial findings.Variations between laboratories and technical approaches, as well as differences in analysis timing and technical details of experimental models, may also affect the final outcomes. It is necessary to evaluate these influencing factors before drawing conclusions or making strategies for intervention. Moreover,despite the difficulty in distinguishing activated microglia/microglia-derived macrophages from infiltrating macrophages based on their morphology and phenotypes in MS studies, it is of great value to reveal their differences in cellular responses and roles since they may contribute differently following demyelination with distinct transcriptional profiles (Yamasaki et al., 2014; Plemel et al., 2020). Microglia-macrophage interactions will provide novel insights into the pathological changes and will establish new strategies to promote remyelination and regeneration.
Summary
Current treatment for MS is mainly focused on immunosuppression or immunomodulation to regulate immune attacks in the CNS. Though a variety of non-curative immunosuppressive therapies are avaible for RRMS, they are unable to prevent disease progression nor revert neurological damges(Wilbanks et al., 2019). The dynamic changes in glial response play vital roles during the progression of demyelination and neurodegeneration, suggesting that glial cells may be innovative targets with great potential in limiting the development of MS. The activated M/M and reactive astrocytes exert both beneficial and detrimental effects on the pathogenesis of MS, dependent on factors lile lesion type and disease course. Thus, therapy strategies should be established based on the pattern and interaction of glial responses. For example, pro-inflamamtory M/M mediate phagocytosis and the clearance of myelin debris is a prerequisite for proper remyelination, but excessive pro-inflamamtory M/M are cytotoxic and exacerbate demyelination and neurodegeneration. Treatment with drugs to stimulate M/M for debris clearance may not be a good idea for progressive MS when persistent and widespread M/M activation exists,while drugs like Tuftsin to shift M/M to an anti-inflammatory/pro-regenerative phenotype may be preferred in this situation. Moreover, as the myelinating cells, oligodendrocyte should be emphasized as a strong candidate for remyelination stimulation. Treatments with chemokines and other molecules to promote OPC recruitment and remyelination are needed after pro-phagocytosis or anti-inflammation treatment. This cocktail therapeutic strategy may maximize the remyelination efficiency considering that an unbalanced lesion environment is hostile to the survival and differentiation of oligodendrocyte linage cells. In addition, cell-type specific treatment may be a favorable direction due to the development of noval drug or siRNA delievery methods. This allows targeted intervention on the response of certain glial cells at the right timing and avoid contrast/unwanted effects on other cell types.
Author contributions:TC, CBS, and JC conceptualized and designed the review. TC and WXZ conducted a literature search. YPZ, YW, and GNB reviewed the clinical relevance. TC wrote the manuscript. LBES, CBS and JC provided substantial edits and improvements of the manuscript. All authors contributed to critical comments and manuscript revision. All authors approved the final manuscript.
Conflicts of interest:The authors declare no conflicts of interest.
Financial support:This work was partially supported by grants from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health (R21 NS098170, to JC and CBS), Kentucky Spinal Cord and Head Injury Research Trust (16-3A, to JC and CBS), and the National Natural Science Foundation of China (81601957, to YW).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix,tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Tetsuya Akaishi, Tohoku University Graduate School of Medicine, Japan; Anna Maria Colangelo, University of Milano-Bicocca,Italy; Olga Chechneva, University of California, Davis, USA.
Additional file:Open peer review reports 1–3.

- 中国神经再生研究(英文版)的其它文章
- Clusterin: a multifaceted protein in the brain
- Positive effects of music therapist’s selected auditory stimulation on the autonomic nervous system of patients with disorder of consciousness: a randomized controlled trial
- Transcranial pulse current stimulation improves the locomotor function in a rat model of stroke
- Comparative transcriptomic analysis of rat versus mouse cerebral cortex after traumatic brain injury
- Delayed atomoxetine or fluoxetine treatment coupled with limited voluntary running promotes motor recovery in mice after ischemic stroke
- Extremely low frequency electromagnetic fields promote cognitive function and hippocampal neurogenesis of rats with cerebral ischemia