
槲皮素分子印迹聚合物的制备及其在处理粮食黄曲霉毒素中的应用
2021-11-15 07:42:40宋立新梁雨涛张云霞王慧格许鹏飞谷立峰
河南工业大学学报(自然科学版) 2021年5期
宋立新,梁雨涛,张云霞,王慧格,许鹏飞,何 娟*,谷立峰
1.河南水利与环境职业学院,河南 郑州 450001
2.河南工业大学 化学化工学院,河南 郑州 450001
3.河南省科学院生物研究所有限责任公司,河南 郑州 450000
谷物在生长、收割、晾晒、储存的过程中易受到真菌毒素的污染[1],其中黄曲霉毒素毒性极大,黄曲霉毒素B1被划分为Ⅰ类天然致癌物[2]。因此,从复杂的样品基质中有效地富集、分离并测定黄曲霉毒素的含量,是保障食品安全至关重要的环节。
目前针对黄曲霉毒素的检测主要采用高效液相色谱法[3-5],GB 5009.22—2016《食品中黄曲霉毒素B族和G 族的测定》对食品中黄曲霉毒素的样品前处理使用的是免疫亲和柱。免疫亲和柱虽然特异性好,但是价格昂贵、只能一次性使用且储存条件苛刻,因此,寻找一种替代免疫亲和柱的材料成为研究重点。分子印迹技术是近些年发展起来的,能够选择性识别某一物质(目标分子)的一种富集技术[6-8]。该技术通过聚合反应合成的分子印迹聚合物(molecularly imprinted polymer,MIPs)经洗脱后,保留空间结构和结合位点[9],这些位点与分析物相似,可选择性吸附目标物,从而达到纯化的目的。随着分子印迹技术的迅速发展,被广泛应用于各个领域,分子印迹-固相萃取常常被用在样品前处理技术中[10-14]。
为避免使用价格昂贵、毒性大的黄曲霉毒素对操作人员的伤害,以及模板洗脱不完全所造成的误差,可选择与黄曲霉毒素结构类似但毒性小的槲皮素作为替代模板[15-17]。作者采用单因素和正交试验对沉淀聚合法制备槲皮素分子印迹聚合物的合成条件进行优化。通过红外光谱、扫描电镜和粒度分布对聚合物的化学组成、外观形态和粒径尺寸进行表征;通过等温吸附和吸附量试验支撑优化结果,测定聚合物吸附性能。将聚合物作为吸附剂制备成固相萃取柱分离和富集发霉小麦中的黄曲霉毒素,分析小麦的霉变情况。
1 材料与方法
1.1 试剂
槲皮素(分析纯)、丙烯酰胺(分析纯)、甲醇(色谱纯):上海麦克林生化科技公司;黄曲霉毒素(AFG1、AFG2、AFB1、AFB2)、乙二醇二甲基丙烯酸酯(EDMA)、偶氮二异丁腈(AIBN):分析纯,上海阿拉丁生化科技股份有限公司;冰乙酸、甲醇、乙醇:分析纯,洛阳昊华化学试剂有限公司。
1.2 仪器与设备
UV-2450紫外-可见光谱仪:日本岛津公司;Nicolet Avatar 360红外傅里叶红外变换光谱仪:美国热电公司;Sigma 300扫描电子显微镜:德国蔡司公司;Mastersizer 2000激光粒度仪:英国马尔文公司; HPLC-2695高效液相色谱仪:美国沃特世公司;DF-101D集热式恒温加热磁力搅拌器:河南省巩义予华仪器有限责任公司;电热恒温水浴锅:北京永光明医疗仪器有限公司;SHB-Ⅲ循环水式多用真空泵:郑州长城科工贸有限公司。
1.3 单因素和正交试验
将模板与单体物质的量之比(1∶ 5、1∶ 6、1∶ 7、1∶ 8)、模板与交联剂物质的量之比(1∶ 15、1∶ 20、1∶ 25、1∶ 30)、引发剂用量(1%、2%、3%、4%)、反应温度(80、84、88、92 ℃)、反应时间(4、5、6、7 h)等作为考察因素,进行单因素试验。
在单因素试验基础上设计模板与单体物质的量之比(A)、模板与交联剂物质的量之比(B)、引发剂用量(C)、反应时间(D)四因素三水平正交试验,对合成反应条件进行优化。
1.4 槲皮素分子印迹聚合物的制备
称取0.604 0 g(2 mmol)槲皮素、0.861 8 g(12 mmol)丙烯酰胺,加入20 mL乙醇作为反应溶剂于50 mL锥形瓶中。超声溶解15 min后,使其充分混合。然后加入7.54 mL(40 mmol)的EDMA、0.263 7 g的AIBN,封口,超声处理5 min,使其全部溶解,将锥形瓶中的体系转入250 mL三口烧瓶中,并添加130 mL乙醇溶剂,加磁子,于88 ℃油浴锅中反应5 h。抽滤,得黄色粉末物质,即为槲皮素分子印迹聚合物(MIPs)。合成后,MIPs需经洗脱将模板分子除去,洗脱液为甲醇与乙酸(体积比4∶ 1)混合液。
以同样的合成方法与条件,在不添加槲皮素的情况下合成非印迹聚合物(non-molecularly imprinted polymers,NIPs)。
1.5 槲皮素分子印迹聚合物的表征
将制得的槲皮素分子印迹聚合物分别按傅里叶红外变换光谱仪要求的KBr压片法、扫描电子显微镜要求的喷金法、激光粒度仪的要求制样后,进行性能表征。
1.6 MIPs和NIPs吸附量的测定
配制不同质量浓度(0.5、1、5、10、15、20、25、30、35、40 μg/mL)的槲皮素溶液,分别取6 mL于10个样品管中,编号,向每个样品管中加入10 mg MIPs,涡旋约15 s,静置1 h后,过滤上清液,计算聚合物对模板分子的吸附量。按照以上步骤对NIPs的吸附量进行测定。
式中:Q为吸附量,μg/mg;C0为吸附前质量浓度,μg/mL;Cn为吸附后质量浓度,μg/mL;V为吸附溶液的体积,mL;m为质量聚合物(MIPs或NIPs)的质量,mg。
1.7 吸附量随时间的变化
配制100 mL质量浓度为30 μg/mL的槲皮素溶液,取10个样品管,每个样品管中加入6 mL槲皮素溶液,再分别加入10 mg MIPs,摇动混匀,过滤,待测。这10个样品液测定的时间分别为0.4、0.5、1、2、3、5、10、20、40 min,计算每个时间的吸附量,绘制吸附量随时间的变化曲线。
1.8 SPE柱制备及使用
采用湿法装柱法自制SPE小柱,100 mg的印迹聚合物放入含有3 mL甲醇的烧杯中,搅拌均匀,然后将其转移到空的固相萃取柱中,固相萃取柱的两端使用聚乙烯筛板进行密封。柱子制备成功后,使用5 mL甲醇活化,并用10 mL去离子水进行洗涤,待用。
参考GB/T 5009.22—2016并稍加修改。将15 mL提取液在自制SPE柱上样,随后用10 mL去离子水洗涤杂质。用2 mL洗脱溶剂(V甲醇∶V去离子水=9∶ 1),洗脱保留在SPE柱中的AFs,控制流速为2滴/s。用N2将洗脱液吹至接近干燥,然后溶于1 mL色谱级甲醇中,0.22 μm滤膜过滤,收集滤液于进样瓶中以备液相色谱进样。
1.9 高效液相色谱法
液相色谱条件:色谱柱为C18柱(150 mm×4.6 mm×5 μm);流动相为甲醇-水(45∶ 55,V/V);流速为0.8 mL/min;进样体积10 μL;柱温25 ℃;检测器为荧光检测器,激发波长360 nm,发射波长440 nm。
1.10 实际样品分析
称取25 g小麦样品于250 mL锥形瓶中,加入100 mL的提取液甲醇-水(V甲醇∶V水=7∶ 3),使用摇床振荡20 min。用普通滤纸进行过滤,再移取滤液按8∶ 42进行稀释,稀释液采用1%吐温-20 和磷酸盐缓冲液(10 mL的吐温-20加入1 L 磷酸盐缓冲液(pH=7.0,8 g NaCl,1.44 g磷酸氢二钠,0.24 g磷酸二氢钾,0.2 g氯化钾,用990 mL的去离子水溶解,浓盐酸调节pH值,最后稀释至1 L))中。最后采用玻璃纤维滤纸过滤,备用。
2 结果与讨论
2.1 单因素试验
在聚合物制备过程中,通过超声处理使单体(丙烯酰胺)与模板分子(槲皮素)以氢键的方式进行预聚合,在交联剂和引发剂的作用下得到聚合物。然后利用有一定弱酸性的混合溶液将模板与单体间的氢键断开,将模板分子洗去,此时的MIPs上保留了模板分子留下的空腔。该空腔在遇到与模板分子结构相似的物质时,可以实现对该物质的吸附。因此,可以通过对模板分子的结构进行选择,从而实现对目标物分离和富集的目标。
根据前人的研究成果[18-20],将模板与单体的比例、模板与交联剂的比例、引发剂用量、反应温度、反应时间等作为考察因素,进行单因素试验。反应温度主要是影响聚合物的转化率,对聚合物的吸附性能影响不大,所以优化后选择合适的反应温度88 ℃。通过单因素试验得到每个量的最佳值:A模板与单体比例1∶ 6、B模板与交联剂比例1∶ 20、C引发剂用量3%、D反应时间5 h。
2.2 正交试验
在单因素试验基础上设计四因素三水平的正交试验,以聚合物的吸附量为指标,对反应条件进行优化。正交试验设计与结果见表1。
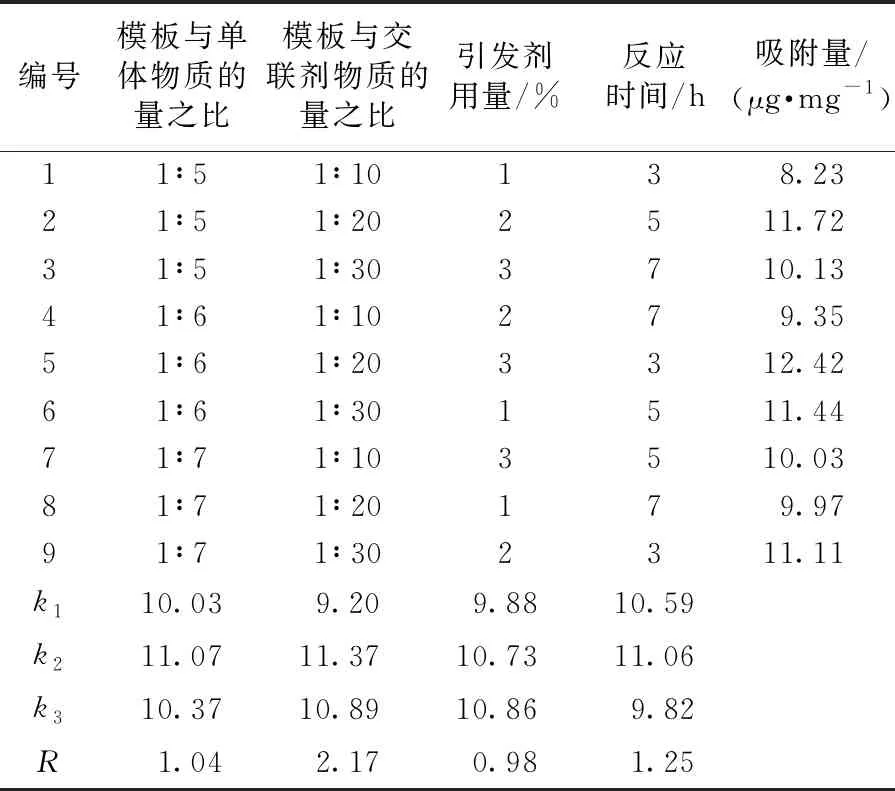
表1 正交试验设计与结果
由表1可知,极差RB>RD>RA>RC,可以判断在聚合过程中,模板与交联剂物质的量之比对聚合物吸附性能的影响最大,其次是反应时间,引发剂用量对聚合物吸附性能的影响最小。因此,通过正交试验得到的聚合物合成优化结果为模板、单体、交联剂的物质的量之比为1∶ 6∶ 20,引发剂用量3%,反应聚合时间5 h。
2.3 聚合物的表征
2.3.1 红外光谱

图1 MIPs的红外光谱图
2.3.2 电镜扫描
由图2可知:聚合物由规则的球形颗粒团聚组成,聚合物微球的粒径大小均匀、合适且分布情况较为均匀。反应得到的产物极具沉淀聚合方法的特点,聚合物的粒径均匀、洁净。此外,聚合物形貌清晰,表面呈现出疏松多孔的特征。聚合物有用作吸附材料的潜力。

图2 MIPs扫描电镜图
2.3.3 粒度分析
如图3所示,聚合物的粒度主要分布在1~10 μm,平均粒度为4 μm,粒径分布均匀。聚合物颗粒在小于1 μm的范围内有分布是在聚合过程中反应不完全引起的。为避免这些颗粒的存在对试验造成的误差,在后续试验中合成的聚合物均经过了轻度的漂洗,利用颗粒在水中的下沉速度不同进行分离,弃去了产物中较细的颗粒。粒度分析结果与扫描电镜结果相吻合,为印迹聚合物能够作为固相萃取柱的填料奠定了基础。
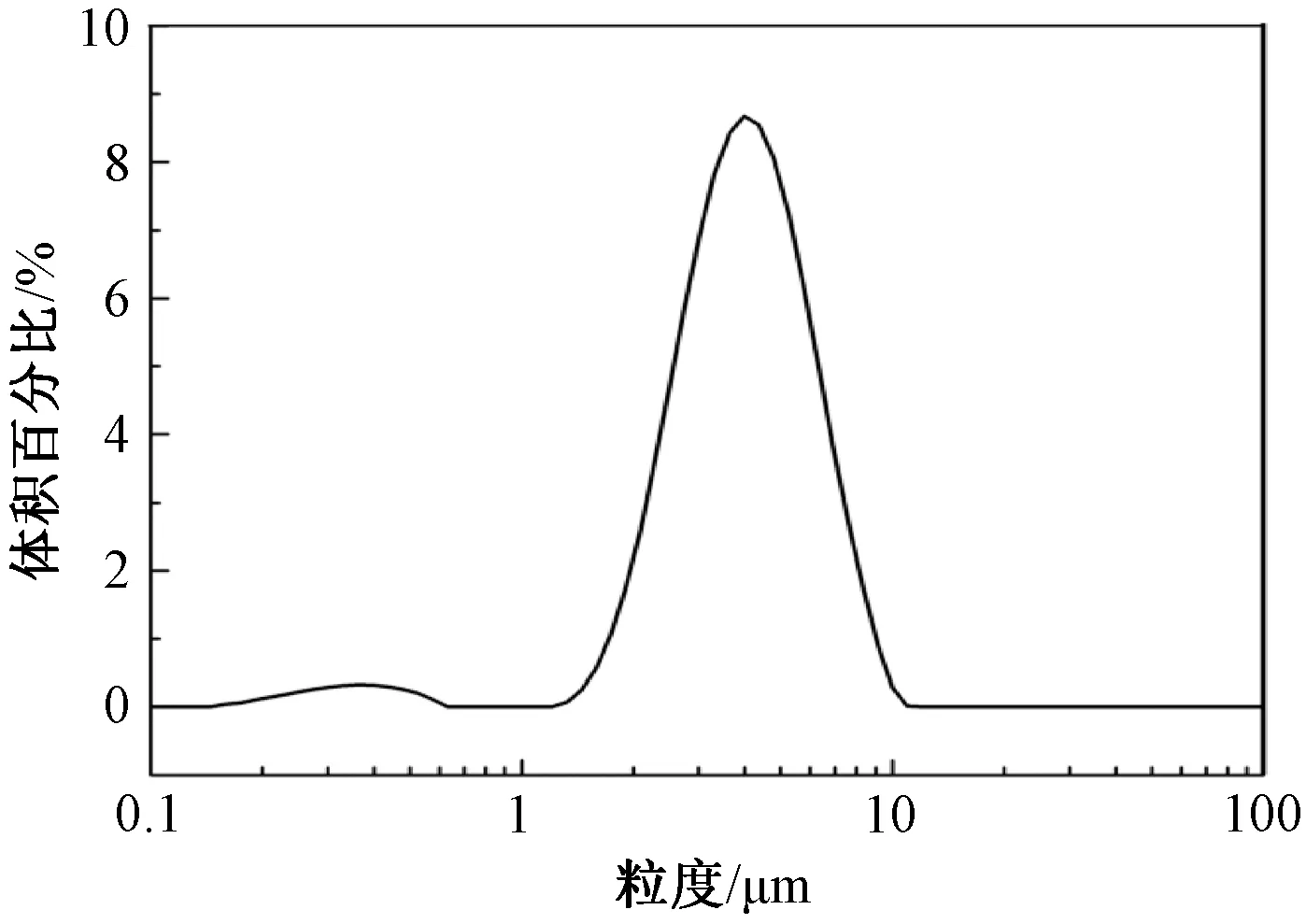
图3 聚合物粒径分布
2.4 吸附性能
2.4.1 等温吸附
图4为MIPs与NIPs分别对不同质量浓度的槲皮素溶液的等温吸附结果,MIPs吸附能力在各个质量浓度的槲皮素溶液中均优于NIPs,且随着溶液质量浓度的增加吸附量持续增大,符合吸附材料应有的吸附性能。随着槲皮素溶液质量浓度的增加,MIPs的吸附性能相较NIPs更加优越。MIPs和NIPs的吸附量随着槲皮素溶液质量浓度的增加都呈上升趋势,且槲皮素质量浓度为40 μg/mL时MIPs吸附量可达到12.76 μg/mg。
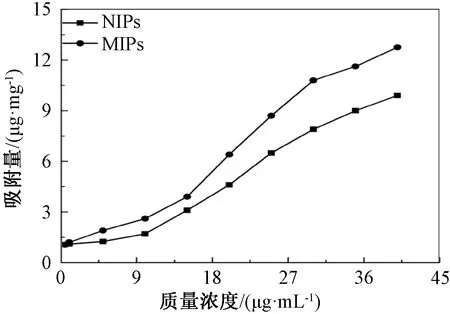
图4 MIPs和NIPs的等温吸附
2.4.2 吸附量随时间的变化
聚合物在相同质量浓度的槲皮素溶液中吸附量随时间的变化如图5所示。聚合物在吸附前期(0~10 min),吸附量变化非常快,随着吸附时间的延长,聚合物对槲皮素的吸附量的增量明显减缓。吸附时间延长到10 min以后,聚合物对槲皮素的吸附量进入平稳期,即吸附已经达到了饱和。在该情况下,增加吸附时间对聚合物的吸附量影响不大,因此,在后续试验中将吸附时间控制在10 min。
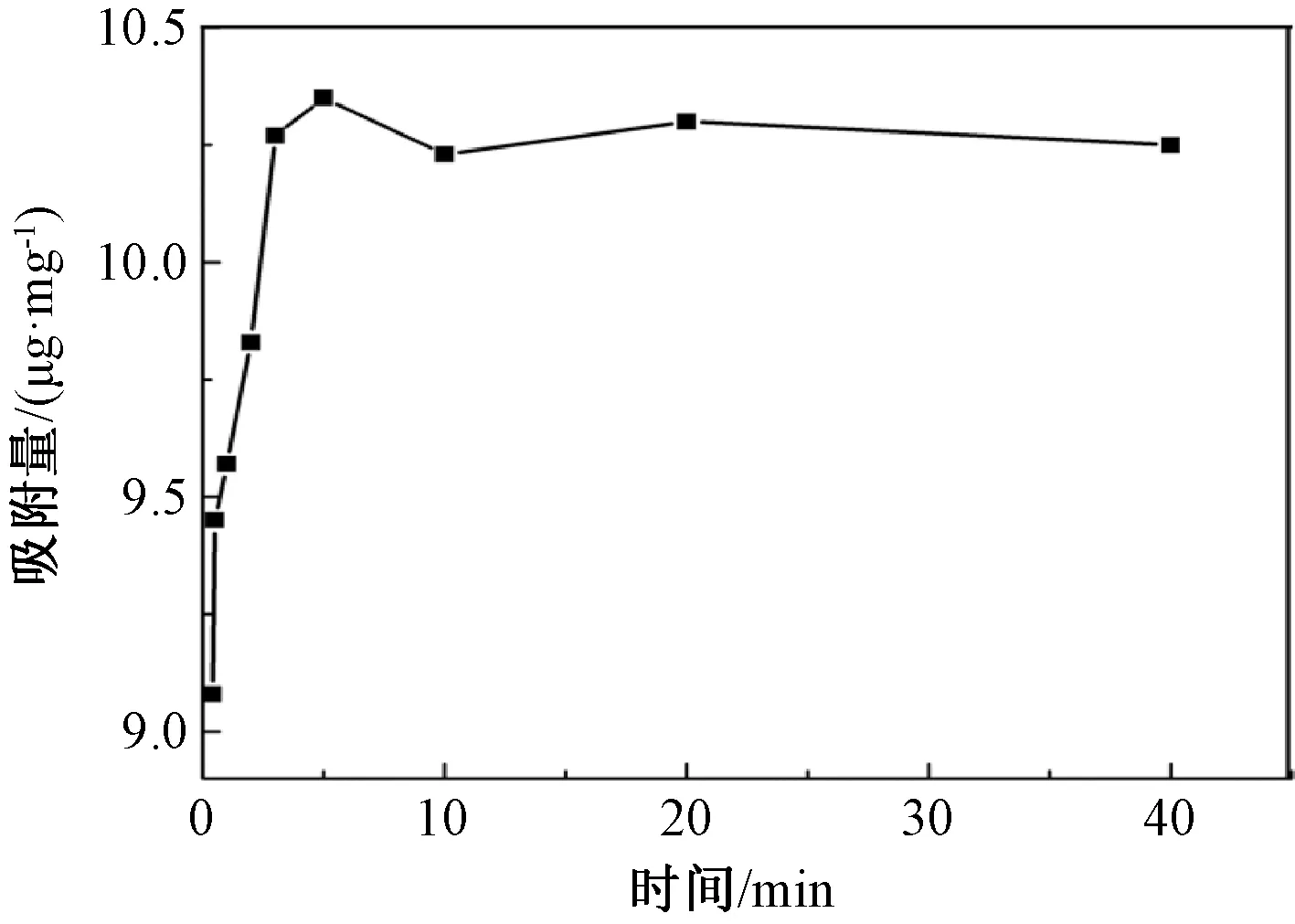
图5 吸附量随时间的变化
2.5 方法评价
以制得的分子印迹固相萃取柱对样品进行前处理,结合高效液相色谱建立方法,为验证该方法的准确性与稳定性,对方法的检出限、定量限、线性范围、准确度和精密度等性能参数进行了评估。如表2所示,4种AFs的标准曲线线性范围为0.5~30 μg/kg,R2=0.999 1~0.999 6,线性良好,可用于对AFs的含量进行定量计算。AFG2、AFG1、AFB2和AFB1的检出限分别为0.05、0.02、0.03、0.05 μg/kg,定量限分别为0.18、0.07、0.10、0.18 μg/kg。检出限和定量限都远低于国家对食品中AFs的限量,自制SPE柱结合高效液相色谱-荧光检测器的方法可用于粮食中AFs的检测。

表2 4种AFs检测方法的检出限、定量限和线性范围
为验证方法的可靠性,根据优化好的SPE柱的固相萃取条件,设定加标量5、10、25 μg/kg,进行加标回收试验,并计算RSD。
由表3和表4可知,在加标量5、10、25 μg/kg条件下,4种AFs的加标回收率为92.5%~118.4%,RSD为3.14%~6.03%。RSD均小于国标中规定的15%,说明自制的SPE柱用于粮食中4种AFs的检测具有较好的回收率和准确度,可用于实际样品中AFG2、AFG1、AFB2和AFB1的分析检测。

表3 4种AFs的加标回收率

表4 方法的精密度
2.6 实际样品分析
将自制的分子印迹固相萃取柱对放置1 a以上的小麦样品中的黄曲霉毒素进行分离和纯化,然后利用高效液相色谱法检测洗脱液中黄曲霉毒素的质量浓度,分析小麦的霉变情况。
对小麦中黄曲霉毒素进行分析,结果如图6

图6 小麦中黄曲霉毒素分析
所示。此时小麦中产生的毒素分别为AFG2、AFG1和AFB2,未产生AFB1。且AFG2、AFG1和AFB2的量分别为33.36、108.66、23.52 μg/kg。在实际样品分析中,发现由于样品放置环境和小麦自身的形态不同,小麦霉变的速度以及产生毒素的含量和霉变位置先后顺序也各有差异。小麦表皮相对较厚,在放置过程前期不易发霉,后期一旦发霉,其霉变速度相比较前期会加快。
3 结论
沉淀聚合法制备MIPs的最佳条件为模板、单体、交联剂的物质的量之比为1∶6∶ 20,引发剂用量3%,反应温度88 ℃,反应时间5 h。得到的MIPs通过红外光谱仪、扫描电子显微镜和粒径分布仪进行表征,结果显示成功合成分子印迹聚合物。MIPs粒径均匀、吸附性能好,优化条件下最大吸附量可达到12.76 μg/mg。以制得的MIPs为填料制备的固相萃取柱对实际样品进行前处理,结合高效液相色谱建立分析方法,并对方法的线性范围、准确度和精密度等性能参数进行了评价。该方法能够用于粮食中痕量黄曲霉毒素的分离、纯化和分析,为监测粮食污染提供了一种新方法。
猜你喜欢
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
中成药(2021年5期)2021-07-21 08:38:40
粘接(2021年2期)2021-06-10 01:08:11
现代畜牧科技(2021年2期)2021-03-19 07:49:22
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
石油化工(2015年9期)2015-08-15 00:43:05
橡塑技术与装备(2015年7期)2015-07-03 12:18:01
中国摄影(2014年12期)2015-01-27 13:57:04
云南畜牧兽医(2014年4期)2014-02-28 21:25:35
