
Alternative RNA splicing in stem cells and cancer stem cells:Importance of transcript-based expression analysis
2021-11-02 00:21EsmaeilEbrahimieSamiraRahimiradMohammadrezaTahsiliManijehMohammadiDehcheshmeh
World Journal of Stem Cells 2021年10期
Esmaeil Ebrahimie, Samira Rahimirad, Mohammadreza Tahsili, Manijeh Mohammadi-Dehcheshmeh
Esmaeil Ebrahimie, Manijeh Mohammadi-Dehcheshmeh, School of Animal and Veterinary Sciences, The University of Adelaide, Adelaide 5005, South Australia, Australia
Esmaeil Ebrahimie, La Trobe Genomics Research Platform, School of Life Sciences, College of Science, Health and Engineering, La Trobe University, Melbourne 3086, Australia
Esmaeil Ebrahimie, School of Biosciences, The University of Melbourne, Melbourne 3010,Australia
Samira Rahimirad, Department of Medical Genetics, National Institute of Genetic Engineering and Biotechnology, Tehran 1497716316, Iran
Samira Rahimirad, Division of Urology, Department of Surgery, McGill University and the Research Institute of the McGill University Health Centre, Montreal H4A 3J1, Quebec, Canada
Mohammadreza Tahsili, Department of Biology, University of Qom, Qom 3716146611, Iran
Abstract Alternative ribonucleic acid (RNA) splicing can lead to the assembly of different protein isoforms with distinctive functions. The outcome of alternative splicing(AS) can result in a complete loss of function or the acquisition of new functions.There is a gap in knowledge of abnormal RNA splice variants promoting cancer stem cells (CSCs), and their prospective contribution in cancer progression. AS directly regulates the self-renewal features of stem cells (SCs) and stem-like cancer cells. Notably, octamer-binding transcription factor 4A spliced variant of octamerbinding transcription factor 4 contributes to maintaining stemness properties in both SCs and CSCs. The epithelial to mesenchymal transition pathway regulates the AS events in CSCs to maintain stemness. The alternative spliced variants of CSCs markers, including cluster of differentiation 44, aldehyde dehydrogenase,and doublecortin-like kinase, α6β1 integrin, have pivotal roles in increasing selfrenewal properties and maintaining the pluripotency of CSCs. Various splicing analysis tools are considered in this study. LeafCutter software can be considered as the best tool for differential splicing analysis and identification of the type of splicing events. Additionally, LeafCutter can be used for efficient mapping splicing quantitative trait loci. Altogether, the accumulating evidence re-enforces the fact that gene and protein expression need to be investigated in parallel with alternative splice variants.
Key Words: Alternative splicing; Stem cell; Cancer stem cell; Transcriptome; Expression analysis
INTRODUCTION
Alternative ribonucleic acid (RNA) splicing is an emerging topic in molecular and clinical studies[1]. Alternative splicing (AS) is the key mechanism to generate a large number of messenger RNA (mRNA) transcripts from the relatively low number of human genes, which can lead to the assembly of different protein isoforms with distinct functions. This structural modification of gene transcripts and their encoded proteins is considered a vital process that increases diversity of protein functions in order to generate the complex cellular proteome[2,3].
Stem cells (SCs) are undifferentiated cells that are able to self-renewal, or differentiate into any types of differentiated cells. SCs can be found in both embryos and adult cells[4]. The self-renewal characteristics also can be found in cancer SCs (CSCs)within the tumor environment[5].
In this review, we discuss the significance of AS in determining the final fate of SCs.Also, the impact of AS events in promoting stemness features in CSCs is highlighted.To address all aspects related to AS, this review provides comprehensive detail regarding various types of AS tools.
MAIN TEXT
Alternative RNA splicing and its different types
The outcome of AS can result in a complete loss of function or the acquisition of new functions[2,3]. AS can also change the gene expression pattern in cancer cells. Exonskipping (a form of alternative RNA splicing) in tumour suppressor genes can lead to truncated proteins, similar to classical nonsense mutations, resulting in cancer-specific AS in the absence of genomic mutations[6]. It has been demonstrated that nearly half of all active AS events are altered in ovarian and breast tumour cells compared to normal tissue[3].
There is ample evidence that AS coordinates significant changes in protein isoform expression and is the main cause of the functional diversity in proteins and proteome[7-10]. In humans, it is estimated that up to 94% of genes undergo AS, resulting in more than 100000 transcripts[7-9].
As presented in Figure 1, transcripts are products of precursor mRNAs (premRNAs) splicing processes, where novel transcripts are discovered with increasing regularity and added to public databases, providing a valuable resource for analysis of AS[11]. During the transcription process, pre-mRNAs are produced. Then, through the RNA splicing process, the non-coding regions of pre-mRNAs (introns) are removed,and coding regions (exons) are joined, and they produce mature mRNAs[12]. Also, it is well-established that the more complex process (AS) may increase the diversity of the mRNAs which are expressed from a single gene. During AS, different exons can be extended or skipped, or introns can be retained within spliced transcripts and produce different mRNAs[13]. The majority (approximately ninety percent) of human genes are alternatively spliced[14]. AS has been considered as a gene expression regulator and miss-splicing can contribute to risk for various human diseases, like cancer[14]. With the advent of high-throughput sequencing methods, analysis of human genomic and transcriptomic has been efficiently developed. Using bioinformatics tools, the sequenced transcripts can be aligned to the genomic reference sequences to find AS events[15,16]. Five standard forms of AS events have been identified including skipped exon (SE, also known as cassette exons), alternative 5’ splice site (A5SS),alternative 3’ splice site (A3SS), retained intron (RI) or intron retention (IR), mutually exclusive exons (MXE). Also, alternative first exons (AFE), and alternative last exons(ALE) are considered as less common AS events[17].

Figure 1 Different types of alternative RNA splicing events.
Exon skipping is the most common AS event, in which the exon as a whole is skipped from the mature mRNA transcript[18]. During A3SS and A5SS events, exons are flanked on one side by a constitutive splice site (fixated) and on the opposite side are flanked by two (or more) competing for alternative splice sites, leading to an alternate region (extension) that either is included within the transcript or is excluded[17]. IR and MXE are the least abundant subtypes among all five major AS events.Normally, during pre-mRNA splicing, introns are fully spliced and excluded from the mature mRNA. However, during IR splicing, introns are retained in mature mRNAs.IR-spliced isoforms have different destinations, some of them may degrade by the nonsense-mediated decay (NMD) pathway, due to the existence of premature termination codons within introns. In some cases, further splicing is applied to the IR isoforms and helps them to escape NMD. NMD escaped-IR isoforms are translated into truncated proteins which have fewer or extra domains in their structure and increase the risk of diseases[19]. In the splicing of MXEs, exons are spliced in a coordinated manner. MXEs can leave the size of the spliced isoform products unchanged. In these isoforms, 1 out of 2 exons (or 1 group out of 2 exon groups) is retained, while the other one is spliced out[20].
There are more events that are not categorised among five standard forms including alternative AFE (alternative promoters) and ALE (alternative 3′ terminal exons). An AFE is the first exon of one splice variant of a gene, which is located downstream of some isoforms of this gene, or this exon is excluded from another isoform because it is located in the intronic region[21]. The AFE definition is also referred to as ALE in which the last exon (3′ terminal exon) may be retained or be excluded in different variants of a single gene[22].
The challenge of alternative RNA splicing analysis in cancer research
There is a gap in knowledge of abnormal estrogen-associated RNA splice variants in breast cancer, and their prospective contribution to breast cancer progression while breast cancer is mainly a disease in which the sex hormone estrogen stimulates uncontrolled growth. Furthermore, their relative abundance and their potential to use AS in cancer diagnosis and treatment has been neglected.
In addition to the novelty of the subject, the major reasons for this shortcoming are:(1) Heavy computation and high computer skills are required for AS detection and analysis; and (2) A high depth of sequencing is required for comprehensive profiling of splicing events. Recently published in Scientific Reports (2018)[11], we developed a Windows-based, user-friendly tool for identifying AS events without the need for advanced computer skills. Additionally, this tool operates as an online module, and employs the SpliceGraphs module without the need for additional resource updates.First, SpliceGraph generates data based on the frequency of active splice sites in the pre-mRNA. Then, the presented approach compares the query transcript exons to SpliceGraph exons in the online genome browser ENSEMBL.
Our team in the Dame Roma Mitchell Cancer Research Laboratories is exceptionally well positioned to unravel the complexity of AS in breast cancer, and thereby, utilise the aberrant alternative splice variants unique to breast cancer as an accurate diagnosis tool. Moreover, this proposed project has the capacity to employ alternative splice variants as a determinant of better outcomes of disease—a completely novel paradigm of disease prevention and diagnosis.
SCs and CSCs
There are various types of SCs. The highest differentiation potential which allows SCs to differentiate into any cell type of a whole organism is found in totipotent SCs, like a zygote. totipotent SCs can generate embryo and extra-embryonic structures in cells[23]. Pluripotent SCs (PSCs) are the other types of SCs that are not able to form extraembryonic structures in cells, but they have the potential to differentiate into cells of three germ layers (endoderm, ectoderm, and mesoderm)[24]. Embryonic SCs (ESCs)and induced PSCs (iPSCs) are categorised in PSCs. ESCs are derived from the inner cell mass of a blastocyst (preimplantation embryos) and the indefinite self-renewal ability and plasticity are their vivid characteristics[25]. iPSCs are artificially derived from somatic cells, and their function and features are similar to PSCs. iPSCs have shown promising impacts on present and future regenerative medicine[26]. Another SC types are multipotent SCs which have limited differentiation abilities than PSCs and they only differentiate into a specific cell lineage. Haematopoietic SCs (HSCs) are multipotent SCs which can only differentiate into blood cells. Unipotent SCs have the least differentiation capabilities which they can only form one cell type, like dermatocytes[27].
Due to the similarities between cancer state and embryonic development, several studies have focused on the existence of CSCs within the tumor environment[5]. It has been well-established that tumor progression, anti-tumor drug resistance, and posttreatment tumor regeneration are driven by a special cancerous cell type, called CSCs.Generally, CSCs characterized by self-renewing, multipotent, and tumor-initiating properties. It is really difficult to detect CSCs within a tumor environment because some CSCs that lack specific markers have tumor regeneration abilities[28]. To illustrate, cluster of differentiation (CD) 133 marker firstly was utilized to isolate CSCs from colon carcinoma, but someCD133-metastatic colon carcinoma cells were found which had self-renewal properties as well asCD133+CSCs[29]. Then, studies showed that CSCs could contain some specific subpopulation, likeCD133-metastatic colon carcinoma cells, they are known as migrating SCs subpopulation[30]. So, in several solid tumors, the subpopulation of migrating and resident SCs can be found which they have been identified by various surface markers includingCD24, CD29,CD44,CD90,CD133, aldehyde dehydrogenase 1 (ALDH1), and epithelial-specific antigen.These markers can be used as a potential target for developing anti-CSCs specific therapies[31].
AS in SCs
The differentiation of SCs and their progenitors relies on various molecular controls associated with the gene expression regulation, including chromatin modification,transcription factors, post-transcriptional regulation by AS, microRNAs (miRNAs),and post-translational modifications. Advanced technologies, as an illustration of high-throughput RNA sequencing (RNA-seq), have demonstrated the contribution of AS patterns in SCs maintenance and differentiation[32].
ESCs are pluripotent cells which able to self-renew, and they have the ability to differentiate into the endoderm, the ectoderm, and the mesoderm cells[33]. Hence,they are the best model for monitoring early embryonic development. Also, ESCs are the potential source for developing differentiated cells for therapeutic approaches in regenerative medicine. One of the first genome-wide studies on splicing patterns in the ESCs reported more than thousands of AS events in ESCs[32]. The results of one study revealed that human and mouse ESC-related AS events are mostly found in genes associated with the cytoskeleton (dystonin, adducin 3), plasma membrane (dynamin 2,integrin subunit alpha 6), and kinase activity (calcium/calmodulin dependent serine protein kinase, microtubule affinity regulating kinase 2, and mitogen-activated protein kinase kinase 7)[34].
Both RNA-seq and splicing microarrays studies reported AS events, particularly those associated with changing protein sequence[15]. Also, the gene expression alteration of the proteins that regarded as splicing regulators was detected during developmental stages, suggesting various AS events during the process of differentiation[16]. For example, increasing expression of splicing regulators like muscle blindlike (MBNL1andMBNL2) RNA binding proteins (RBPs) was shown during ESC differentiation which is highly associated with ESC-differential AS. The presence of the MBNL motif downstream and upstream of the flanking intronic sequences is associated with exon skipping and exon inclusion in ESC respectively[34]. It has been revealed that the AS events of genes that they contribute to ESC differentiation foster this process, like embryonic SC-specific event in forkhead box p1 (FOXP1),FOXP1-ES.TheFOXP1-ESspliced isoform stimulates the expression of genes, including octamerbinding transcription factor 4 (OCT4), nanog homeobox (NANOG), nuclear receptor subfamily 5 group A member 2, and growth differentiation factor 3 which these transcription factors are required for pluripotency. Along with maintaining ESC pluripotency,FOXP1-ESshowed effective involvement in the reprogramming of somatic cells to iPSCs[35,36]. The fibroblast growth factor 4 splice isoform (FGF4si) ofFGF4is another example of transcription factor that play major roles in ESC fate determination and promotes differentiation at later stages of development[37].
One of the most important regulators of self-renewal in ESCs is POU domain proteins (POU class 5 homeobox 1, also known asOCT4) and its gene expression is stimulated byFOXP1-ES.OCT4has various isoforms includingOCT4A,OCT4B,OCT4B1,OCT4B2, andOCT4B4. The expression ofOCT4Aonly has been detected in ESCs and embryonal carcinoma cells (ECCs). TheOCT4Bdetected in differentiated cells andOCT4B1had higher expression levels in ESCs and ECCs rather than nonpluripotent cells[38]. Also, it has been revealed that expression patterns ofOCT4B2andOCT4B4are similar toOCT4A(they highly expressed in undifferentiated cells),and likeOCT4B1, their expression is lower in differentiated cells[39,40].
AS events also have found as critical factors to control the hematopoiesis process which during this process the HSCs produce mature blood cells in the bone marrow.The importance of AS in hematopoiesis has been identified through the analysis of AS related to RUNX family transcription factor 1 (RUNX1) which is a critical transcription factor for this process.RUNX1aisoforms are generated fromRUNX1by exon 6 AS and increase the capacity of the HSC pool[41]. Moreover, multiple isoforms of the other HSC-specific genes (homeobox A9, Meis homeobox 1, PR/SET domain 16, and HLF transcription factor-PAR bZIP family member) have been found using wholetranscriptome splicing of murine HSCs[42]. Also, a bioinformatics analysis revealed that the IR in HSC-specific transcripts led to a decrease in the expression of genes involved in DNA binding and RNA processing. This process consequently promoted the NMD pathway in HSCs[43,44].
Along with ESCs and HSCs, there are neural SCs (NSCs) that are responsible for generating neurons and glia during embryonic development. The nervous system applies AS for cell differentiation, morphogenesis, and the formation of complex neuronal networks. The three major alternatively spliced isoforms of Quaking (Qki)have been found in NSCs.Qki5is one of these isoforms which regulates NSC functions[45]. Another example of AS involvement in the transition of SC to neuron cells is the existence of exon 10 in the mRNA of polypyrimidine tract binding protein 2 (PTBP2)protein[46]. Interestingly, it has been cleared that the downregulation levels of the other paralog of this protein, polypyrimidine tract binding protein 1 (PTBP1), inhibit the existence of exon 10 ofPTBP2[47]. Hence, in neural cells, the neuron-specific microRNA miR-124 contributes to the downregulation ofPTBP1[48]. On the other hand, there is another ubiquitous RNA-binding protein which suppresses PTBP1/PTBP2 levels by inhibiting the inclusion of exon 11/10 ofPTBP1/PTBP2in myoblast cells[49,50].
AS and CSC
SCs in normal tissues are capable of renewing themselves, also, stem-like cells within tumors, which have been called CSCs have the ability of self-renewing to seed new tumors. Hence, they have also been termed “tumor-initiating cells”[51].
Due to gene expression regulation at the transcriptional level, the contribution of AS events has been clearly reported in tumor-related biological processes like proliferation, cell death, migration, and angiogenesis. AS changes transcriptome and proteome profile in human cells, therefore, its deregulation may greatly contribute to tumor plasticity[9]. Effectively, AS plays a regulatory role in maintaining the balance between pluripotency and differentiation of human ESCs during embryogenesis and tissue differentiation. Hence, defective AS machinery could mimic the oncogenic effects in non-tumorigenic cells. Results of an RNA-seq-based study on mammary cells revealed that AS events regulated by serine and arginine rich splicing factor 1 altered in human tumors. These defective AS events led to an enhancement of the proliferation and decreased apoptosis in MCF-10A cell cultures[52]. The involvement of defective AS events in controlling tumor heterogeneity suggesting that they could also lead to re-programming of stem pathways, triggering metastasis, and tumor progression[53].
Phenotypic conversions of cells between epithelial and mesenchymal states, known as epithelial-mesenchymal transition (EMT), is activated during metastasis and enhances the re-activation of stem pathways. Hence, EMT is associated with tumor aggressiveness and resistance of cancer cells to anti-tumor drugs[54]. Various types of regulators including cytokines and growth factors are dysregulated in cancer cells which mainly involved in promoting EMT. EMT influences the mRNA maturation of some splicing factor-like epithelial splicing regulatory protein (ESRP). This splicing factor regulates the Wnt signaling pathway through exon 4 skipping of T-cell factor 4 (TCF4). The activation ofTCF4is prompted by nuclear localization of beta-catenin and they are major transcriptional mediators of the canonical Wnt signaling pathway[55].It has been revealed that, during EMT, the transactivation of exon 4 carryingTCF4isoforms is reduced. On the contrary, the lack of exon 4 Led to surge Wnt signaling during EMT[56].
Moreover, ESRP-splicing factors can alter splicing of the Fibroblast growth factor receptor 2 (FGFR2)[57]. There are two different isoforms ofFGFR2including IIIb and IIIc which have pivotal roles in ligand binding specificity. ESRPs mainly help to the production ofFGFR2-IIIbby inhibiting exon IIIc. TheFGFR2-IIIclevels increase in the absence of ESRPs[58]. Spliced transcripts ofFGFR2were detected in primary tumors,and significantly enriched in metastases and tumor plasticity[59].
Also, the association between EMT and the emergence of CSCs has been identified[60]. Studies showed that EMT induces AS events in genes involved in stem-like cancer cells. NUMB endocytic adaptor protein (NUMB) is an endocytic adaptor protein that has various functions in cell polarity maintenance, cell migration, and EMT. The importance ofNUMBAS has been reported in various cancers including breast[61,62], lung cancer[63], and hepatocellular carcinoma cells[64]. FourNUMBisoforms based on the AS of exon 3 and exon 9 have been identified in vertebrates[65].Exon 9 mainly founds in SCs rather than differentiated cells, also enhanced expression ofNUMBexon 9 has been clearly shown in various cancer types. Interestingly,MEK/ERK signaling pathway regulatedNUMBexon 9 splicing[66]. In cancer cells,EMT triggersNUMBexon skipping, which leads to enhance invasive properties, and abnormal AS events inNUMBmay also affect the balance between stem-like and nonstem cancer cells[67].
One of the most important SC factors which contribute to embryogenesis and pluripotency of cancer stem-like cells is a member ofPOUfamily genes calledOCT4.This SC factor plays a pivotal role in self-renewal and pluripotency properties in ESCs and ECCs[68]. AS leads to produce three main isoforms of the humanOCT4, includingOCT4A,OCT4B, andOCT4B1[38].OCT4Acontributes to maintaining stemness properties in ESCs and ECCs. On the contrary,OCT4Bisoforms are not able to show these features[38,39].
Another gene that its spliced variants have been found in CSCs is Kirsten rat sarcoma viral oncogene homolog (KRAS). The occurrence of mutation and AS in this gene results in the production of transformed proteins that promote malignancies[69].The spliced variants ofKRASareKRAS4AandKRAS4Bwhich are known as minor and major isoforms respectively and in CSCs the splicing process ofKRASis regulated by the RNA binding motif protein 39 splicing complex. Hypoxic state in tumor environment activates the expression ofKRAS4Ain CSCs and there is a correlation between the expression ofKRAS4Aisoform and SC marker ALDH in cells with stemlike properties.KRAS4Aspliced variant controls the metabolic requirements in SCs particularly the level of adenosine triphosphate and lactate. These metabolites increase in the absence ofKRAS4A. Furthermore, Chenet al[70] reported that Indisulam, which is a novel sulfonamide compound with potential antineoplastic activity, is an inhibitor ofKRAS4Asplicing, but has no obvious effect onKRAS4B. Also, previously the involvement ofKras4isoforms in murine SCs during development was reported[71].
Erb-B2 receptor tyrosine kinase 2 (HER2) is a member of the epidermal growth factor receptor family of receptor tyrosine kinases and its overexpression is a common feature of invasive breast carcinomas. Cells that express HER2 receptors considered as HER2-positive breast cancer. The full-length HER2 is known as wild-type HER2(wtHER2) which has about 1255 amino acids. While the altered form of HER2 has been identified with the absence of sixteen amino acids from the extracellular domain(deletion of exon 16). ThisHER2splice variant is known asd16HER2which highly enriched in the regulation of the breast CSCs (BCSCs) activity by its functional interaction with the notch receptor 1 family members[72]. Also,d16HER2spliced variants are involved in initiation and aggressiveness of tumors, CSC properties, EMT,and the trastuzumab susceptibility of HER2 positive BC cells compared with wtHER2[73].
Another example of EMT-promoting splicing changes is occurred by two splicing factors including QKI and RNA-binding Fox proteins (RBFOX1/RBFOX2). QKI and RBFOX2 are responsible for exon skipping of cortactin transcript during EMT[74]. The roles of QKI and RBFOX1 in establishing the SC features in breast tumor cells and regulating the EMT have been declared by applying exon 30 splicing (exon skipping)in the actin-binding protein FLNB[75].
AS in the CSC marker
One of the most well-known cell surface adhesion receptors of CSCs isCD44which also famous as a CSC marker. CD44 is a non-kinase transmembrane glycoprotein comprised of 20 exons which AS leads to produce two different isoforms including the standard (CD44s) and variant (CD44v) isoforms[76]. Exons 1-5 and 16-20 are involved inCD44sisoform and make it the smallest isoform which is expressed by the mesenchymal cells. On the other hand, the middle nine exons (exons 6-15 of the genomic DNA) can be alternatively spliced and located between exons 1-5 domain and exons 16-20 region which formCD44visoforms[77]. CD44 activates by binding to its main ligand, hyaluronic acid. Activated CD44 Leads to cell proliferation and metastasis[78].CD44sisoforms predominantly express on hematopoietic and mesenchymal cells and theCD44sisoforms abundance in cancer cells induces stemlike features[79]. This protein is highly expressed in many cancer cells and its alternative spliced variants play a critical role in tumor progression.
Previously, it has been reported that switching fromCD44vtoCD44sisoform led to promoting EMT in cells. Cancer cells that undergo an EMT acquire SC-like properties,andCD44expression increases in these cells, while the expression levels of splicing factorESRPreduced[54]. Splicing factor ESRP contributes to switchingCD44visoform intoCD44swhich is required for promoting EMT[80]. Various studies have been done to reveal the exact role ofCD44isoforms in cancer cells. It has been reported that increasing tumor cell survival, invasiveness, and migration is associated withCD44s[81]. Ouhtitet al[82] showed the increased expression ofCD44in metastatic breast tumors and they providedin vivoevidence for the role of theCD44sisoform in promoting breast tumor invasion and metastasis to the liver. Moreover, Hiraga etal[83] declared thatCD44can induce functional properties in CSCs and itsCD44svariant contributes to the promotion of bone metastases. BesidesCD44s, there is some evidence that proved the role ofCD44vin enhancing CSC activities[84,85]. Also,protecting CSCs from reactive oxygen species-induced stress by interacting with a glutamate-cystine transporter that subsequently promotes tumor growth[86].
One of the driving factors that control EMT-associated splicing changes are ESRP-1 and -2 splicing factors. ESRP1 inhibits CSC traits by inhibiting the production ofCD44s. Increased expression ofCD44supon EMT-induction leads to the invasive phenotype in cancer cells. Also, the results of bioinformatics analysis of the Cancer Genome Atlas program of breast cancer showed that there is a significant negative correlation between the ratio of theCD44isoforms and theESRP1splicing factor[79].Also, it has been proved that the expression levels ofESRP1decreased in triplenegative breast cancer (TNBC) compared with non-TNBC samples. Along with direct regulatory effects of ESR1 onCD44sisoform, this splicing factor also controls the CSCs functions by splicing α6β1 integrin. α6 integrin subunit (CD49f) is a well-known biomarker for BCSC and other CSCs and there are two splice variants for this subunit including α6Aβ1 and α6Bβ1 isoforms[87,88]. ESRP1 impedes CSC function by inducing the expression of the α6Aβ1 splice variant and repressing α6Bβ1 integrin isoform[58,89].
Despite the above arguments, there are some controversies about the role of ESRP1 in promoting[90] or inhibiting[89] CSC properties. The results of a study which was conducted by Yaeet al[90] seem contradictory. They reported that ESRP1 contributes to breast cancer metastasis through a mechanism in which these splicing factors stimulate the upregulation of theCD44visoforms and results in lung metastasis of breast cancer cells accompanied by the expansion of stem-like cancer cells[90]. Also,Huet al[91] revealed thatCD44vis expressed onCD24-/CD44+breast CSCs which enhances the risk of metastasis to the lung, rather than cells that expressingCD44s.
ALDHis a common CSC marker which catalyses the oxidation of aldehydes. There are 19 various types ofALDHin the human genome. Upregulation ofALDHshas found in SCs and CSCs[92]. However,ALDH1mainly considered as SCs and CSCs markers and contributes in self-renewal activity which has three isoenzymesALDH1A1,ALDH1A2, andALDH1A3. Among these isotypes,ALDH1A1is the most prominent SC marker in renal cell carcinoma (RCC) tumor, breast cancer[93], colon cancer[94], and is linked to tumorigenesis, mortality, and self-renewal activity[95].
Another marker of CSCs in the gastrointestinal tract is doublecortin-like kinase 1 (DCLK1) which mostly correlated with tumor initiation, EMT, and progression[96,97].In RCC tumor,DCLK1alternative spliced variants (DCLK1 ASVs) overexpressed compared to control samples.DCLK1-long isoforms (Isoforms 2 and 4) are associated with RCC recurrence and they mainly co-expressed with renal tumor SC markers includingALDH1A1, C-X-C motif chemokine receptor 4, andCD44. It has been proved that a high level ofDCLK1alternative transcript (Isoform 2) promotes the expression of RCC SC markers and increases self-renewal activity[98].
In summary, the AS machinery could regulate the self-renewal features of stem-like cancer cells. This process is performed by producing spliced variants of cancer cell markers. The most important spliced variants of CSCs markers areCD44s,CD44v, and α6Bβ1 integrin because they have found on the surface of various CSCs. Also, the splicing factor ESRP is responsible for the splicing changes of bothCD44isoforms and α6Bβ1 integrin. The Table 1 represents the association of the alternative RNA splice variants and their activities in both SCs and CSCs[99-105].

Table 1 Genes that undergo splicing events in stem cells and stem-like cancer cells

IGF-1: Insulin like growth factor 1; POU5F1: POU domain proteins; OCT4: Octamer-binding transcription factor 4; RMB: RNA binding motif protein;DNMT3B: DNA methyltransferase 3 beta; VEGFA: Vascular endothelial growth factor A; FGF4: Fibroblast growth factor 4; PKCd: Protein kinase C delta;RUNX1: RUNX family transcription factor 1; Qki: Isoforms of Quaking; PTB: Polypyrimidine tract binding; FOXP1: Forkhead box p1; NUMB: NUMB endocytic adaptor protein; KRAS: Kirsten rat sarcoma viral oncogene homolog; HER: Erb-B2 receptor tyrosine kinase; CD: Cluster of differentiation; ITGA5:Integrin subunit alpha 5; FLNB: Filamin B; DCLK1: Doublecortin-like kinase 1; ALDH1: Aldehyde dehydrogenase 1.
AS analysis tools
Since the pivotal role of AS events has been cleared in increasing self-renewal properties and maintaining the pluripotency of CSCs, it is highly important to detect differential splicing using computational approaches. Sequencing methods have paved the way to survey AS. Initial studies by microarray profiling and EST-cDNA sequence data reported that about two-thirds of human multi-exon genes are alternatively spliced[106]. Then, high-throughput sequencing technologies provide a high depth of coverage and sensitivity to identify human AS. For the first time to survey the splicing complexity, the Genome Analyzer system of Illumina was used by Panet al[7] Their results proved the effectiveness of the RNA-seq method to analyze AS events.However, it has been found that the RNA-seq method is not sufficient itself due to its short sequencing reads (approximately 100-150 bp), and needs a number of computational approaches to monitor differential splicing[107].
There are three approaches to perform differential splicing analysis, including exonbased, isoform-based, and event-based methods. Mainly, exon-based and event-based methods are categorized in one strategy called the count-based method (Table 2)[108-146].
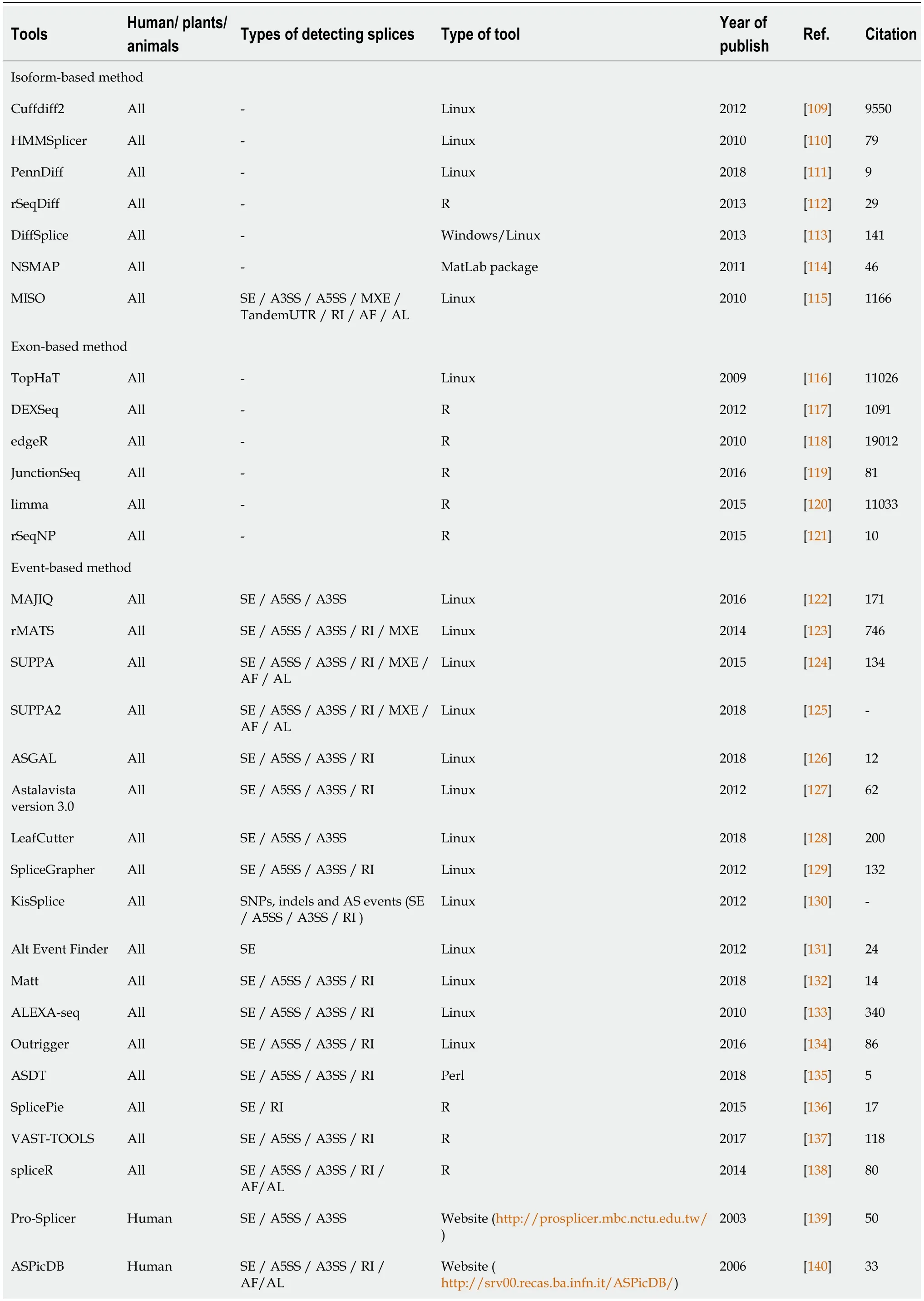
Table 2 Alternative splicing analysis tools based on isoform-based method and count-based method (including the exon- and eventbased approaches)

SE: Skipped exon; A3SS: Alternative acceptor site; A5SS: Alternative donor site; RI: Retained intron; MXE: Mutually exclusive exons; AF/AL: Alternative first/last exons.
Isoform-based method
Isoform-based methods are based on the reconstruction of full-length transcripts at the first step, then these methods estimate the relative isoform abundance across samples or conditions. Following that, statistical testing is used to identify the significant differences in the relative transcript abundances among various experimental conditions and samples. It is noticeable to say that the effectiveness of this method relies on accurate transcript quantification. Some of the isoform-based tools have been developed including cuffdiff2, HMMSplicer, PennDiff, rSeqDiff, DiffSplice, NSMAP,and MISO. The last two mentioned tools are also able to detect splicing events while it is not possible for others.
Cufflinks is an isoform-based pipeline which contains three programs including Cufflinks, Cuffmerge, and Cuffdiff. Cufflinks first applies transcript assembly by generating overlap graphs with fragments and quantifying the aligned reads.Transcript abundance of a transcript is then estimated in form of FPKM (fragments per kilobase per million mapped fragments). Then, using Cuffmerge, collected assemblies are merged to create a consensus reference. Cuffdiff2 finally performs different tests for detecting differentially expressed genes and differential isoform changes are calculated by applying a one-sided t-test[147].
HMMSplicer is a precise algorithm for analyzing canonical and non-canonical splice junctions in short-read datasets. HMMSplicer firstly divides each read in half, then seeds the halves to the reference genome and based on Hidden Markov Model, finds the exon boundary. At final points, a score is assigned to each junction, based on the alignment strength, number, and quality of bases supporting the splice junction. The true and false positives can be distinguished perfectly using this scoring algorithm and lead to find novel both canonical and non-canonical splice junctions[110]. PennDiff, as an accurate statistical method takes information regarding both gene structures and pre-estimated isoform relative abundances into consideration, then analyzes differential AS or transcription for RNA-seq data[111].
rSeqDiff is implemented as an R package for the detection of differential isoform expression from multiple RNA-Seq samples using the hierarchical likelihood ratio test.rSeqDiff can considering three cases for analysis, including genes with no differential expression, genes with differential expression without differential splicing, and genes with differential splicing[112].
DiffSplice is additionally another isoform-based method for the detection and visualization of differential transcription. The DiffSplice approach is not based on transcript or gene annotations, it overcomes the requirement for full transcript inference and quantification, which may be a challenge due to short read length. So,what makes DiffSplice distinct from other methods is that this tool uses a divide-andconquer approach to seek out the difference between transcriptomes within the variety of AS modules (ASMs), where transcript isoforms separate. The abundance of various AS isoforms existing in each ASM is calculated for every sample and is compared across sample groups. A non-parametric statistical test is used for each ASM to demonstrate significant differential transcription with a controlled false discovery rate(FDR)[113].
NSMAP (non-negativity and sparsity constrained maximum a posteriori) model is provided to estimate the expression levels of isoforms using RNA-seq data. Like DiffSplice, NSMAP does not require annotation information. This tool drives the structures of isoforms and estimates the expression levels simultaneously[114].
MISO (mixture of isoforms) model with the help of bayesian inference estimates the expression level of alternatively spliced genes from RNA-seq data. Also, it is a probab-ilistic framework that takes RNA-seq data of single-end or paired-end to perform more accurate AS analysis at either the exon or isoform level. Interestingly, MISO provides confidence intervals for estimates of exon and isoform abundance, detects differential expression, and uses latent information to enhance accuracy. Also, this tool using GFF annotations can generate various types of AS events including SE, A3SS/A5SS, MXE,TandemUTR, RI, AFE, and ALE[115].
Count-based method
Count-based methods comprised of both exon-based and event-based models. In exon-based approaches read counts are assigned to different features, such as exons or junctions. The main difference between these two approaches is that exon-based methods are not able to provide the type of splicing event; they just estimate the differentially expressed exons/junctions between samples. Tools which have been developed based on the exon-based methods are TopHaT, DEXSeq, edgeR, JunctionSeq, limma, and rSeqNP. Except for TopHat, all exon-based tools are launched in the R environment.
TopHat is a software package that finds spliced junctions ab initio by large-scale mapping of RNA-seq reads. TopHat performs mapping the reads using Bowtie (http://bowtie-bio.sourceforge.net), an ultra-fast short-read mapping program to reference genome[116]. Also, TopHat is considered as a prior separately running tool for other software including Alt Event Finder, FineSplice, SplicingCompass, and NSMAP[148]. Firstly, the RNA-Seq data is processed by TopHat to find the splicing junctions. Then, NSMAP re-counts all the possible isoforms formed by the combinations of collected exons from TopHat, and identifies the expressed isoforms and their expression level estimations[114].
DEXSeq is another exon-based method which uses a linear model to detect differential splicing genes from RNA-seq data[117]. Identifying differential expression levels of genes, exons, or transcripts is also done by the edgeR package. Moreover, using a negative binomial generalized log-linear model, edgeR can be used to analyze the count data. Then, the expression levels are calculated by comparing the logFC of an exon to the logFC of the entire gene[118]. The JunctionSeq package works based on the statistical approach of DEXSeq to calculate the differential exon usage and exon junctions[119].
Limma package is a well-known R package for detecting differential gene expression also provides differential splicing using exon count data. Limma integrates various statistical principles to perform large-scale expression studies accurately. First,this package applies a linear model to calculate differential expression tests for the exon-level expression data. Then, the exon-level statistics are turned to gene-level statistics for detecting differential spliced genes[120].
rSeqNP like other exon-based methods is able to test differential expression and differential splicing of genes. This tool uses standard non-parametric tests based on ranks of expression values of genes and isoforms[121].
Event-based methods through calculating the percentage spliced in (PSI) values can generate splicing events including SE, A3SS/A5SS, MXE, RI, AFE, and ALE. There are various types of software in this category which some are available as python, R, Perl packages, or others are accessible through online websites. Some of the most important tools are MAJIQ, rMATS, LeafCutter, SUPPA/SUPPA2, ASGAL, spliceR, SpliceDitector,etc.
MAJIQ (modeling alternative junction inclusion quantification) is able to find splice graphs and local splice variations (LSV) using RNA-seq data and transcriptome annotation file. An LSV can be determined as a split in a splice graph or from a single exon (reference exon). Single source LSV is related to splits from a reference exon to multiple 3’ splice sites in downstream exons, while, single target LSV is related to multiple 5’ splice sites spliced to an upstream reference exon. Both simple splicing events and complex transcript variations can be included in the LSVs. The MAJIQ builder and MAJIQ quantifier make up MAJIQ. The MAJIQ builder tries to detect known and novel LSVs and construct a splice graph for genes collected from RNA-seq data and transcriptome annotation. Following that, the MAIJQ quantifier applies Bayesian PSI modeling and bootstrapping to estimates PSI values for each quantified LSV. MAJIQ is mainly used with the Voila package which is a visualization tool that using the output of MAJIQ (builder and quantifier), creates interactive summary files with gene splice graphs, LSVs, and their quantification[122].
MATS (multivariate analysis of transcript splicing) is a Bayesian statistical framework for the identification of differential spliced genes obtained from RNA-Seq data. MATS applies multivariate uniform before estimating the correlation of exon splicing between samples, also it uses a Markov Chain Monte Carlo (MCMC) method along with a simulation-based adaptive sampling method to report thePvalue and FDR of differential AS[149]. rMATS is a developed version of the MATS method.rMATS uses a hierarchical framework to account for sampling uncertainty in individual replicates and variability among both paired and unpaired replicates between sample groups, also it can estimate the PSI of each event[123].
LeafCutter is an event-based method developed to analyze samples and population variation in intron splicing using short-read RNA-seq data. LeafCutter can detect differential splicing between sample groups, and used for mapping splicing quantitative trait loci (sQTLs). So, analyzing sQTLs by LeafCutter can help to identification of disease-associated variants. To compare LeafCutter and MAJIQ tools,MAJIQ is used to predict local splicing variation using split-reads and identification of complex splicing events, but MAJIQ does not scale properly more than thirty samples and has not been adapted to map sQTLs. The LeafCutter output of AS events is mainly focused on SEs, 5′ and 3′ alternative splice site usage, and additional complex events that can be summarized by differences in intron excision. Comparison of LeafCutter to other methods for differential splicing analysis revealed that the majority of the introns that LeafCutter reported as the most significant differentially spliced, shared a splice site with rMATS and MAJIQ. It can be considered that LeafCutter is able to detect the same differentially spliced events[123].
SUPPA is another event-based method which uses RNA-seq data to calculate PSI for differential spliced events[124]. SUPPA and its different version, SUPPA2, are able to consider AFE and ALE events coupled with other standard types of events. SUPPA2 has more advantages, including working with various replicates per condition and with different conditions. Also, SUPPA2 is able to cluster the differentially spliced events among various conditions to determine common splicing patterns and regulatory mechanisms[125].
Alternative Splicing Graph ALigner (ASGAL) is an event-based tool that uses RNASeq samples and a gene annotation to detect AS events. Firstly, ASGAL constructs the splicing graph from the gene annotations. Then, the RNA-reads alignment is done against the constructed splicing graph of the input gene. Finally, AS events are determined. The most prominent feature of ASGAL is that this tool can have higher accuracy to predict events. Because as compared with other tools which perform spliced alignment against a reference genome, ASGAL tries to detect novel splice sites based on a splicing graph[126].
SpliceR is an R package to detect AS events uses the full-length transcript output from RNA-seq data. For each event, spliceR annotates the genomic coordinates of the differentially spliced elements, to enhance downstream sequence analysis. Moreover,the possibility of the coding potential and NMD sensitivity of each transcript are determined by spliceR[138].
SpliceDetector is a windows-based user-friendly software for the identification of AS events directly from transcripts without any computer skill requirement or database download. Furthermore, to construct splicing graphs, data updating is not necessary because SpliceDetector uses the updated information deposited in the Ensembl database. To use SpliceDetector software, first, it takes human, plant, and model organisms transcript IDs as input. Then, it constructs a SpliceGraph based on all of the exon coordinates of the related gene. Finally, AS events are provided as output[11].
The interaction between epigenetic and AS within SCs and CSCs
The critical role of AS in producing different mRNA variants, and even non-coding RNAs (ncRNAs), form one gene is well-established. AS can directly enhance transcriptomic and proteomic diversity. AS is regulated by RBPs which monitor the splicing machinery by binding to the pre-RNA. AS can be subjected to epigenetic regulation, like DNA methylation and histone modifications, and regulation by ncRNAs[150]. Long ncRNA (lncRNAs) are ncRNAs mainly longer than 200 nucleotides which have emerged as an essential regulator in various cellular processes[151].Also, the critical roles of lncRNAs in adult SCs have been identified. Long intergenic non-protein coding RNA PNKY (Pnky) is a neural-specific lncRNA within the nucleus of NSCs. This lncRNA maintains the undifferentiating features of NSCs and neural differentiation enhanced by knocking downPnky. Interestingly,Pnkycould regulate AS through interacting with splicing regulator PTBP1[152,153].
MiRNA are small non-coding RNAs, about 23 nucleotides in length, play role in gene expression regulation by pairing with complementary sequences in proteincoding transcripts[154]. The microRNA-290 cluster maintains the pluripotency and stemness properties of ESCs by monitoring AS, also known as a master regulator of AS. The splicing factorsMBNL1/2are targeted by one of the members of this cluster,miR-294, and the downregulation levels ofMBNL1/2have been found in ESCs and during reprogramming of iPSCs. Also, it has been reported that the majority of AS events are regulated by miR-294 throughMBNL1/2repression and this repression leads to up-regulation of other splicing factors in ESCs, so miR-294-dependent AS events are enhanced[155].
Also, the indirect involvement of some specific spliced variants in the miRNA expression regulation process has been demonstrated in CSCs. CSCs of human head and neck squamous cell carcinoma (HNSCC) are mostly characterized byCD44v3isoforms which generate through alternative mRNA splicing ofCD44. Also,ALDH1markers along with the high expression of transcription factorsOCT4, SRY-box transcription factor 2 (SOX2), andNANOGare the other features of HNSCCs. In CSCs,Oct4-Sox2-NanogTFs are activated by binding theCD44v3to its ligand hyaluronan and these TFs have binding sites on miR-302 promoter. So, the interaction between hyaluronan-inducedCD44v3-spliced variants and mentioned TFs plays a pivotal role in the downregulation of miR-302 target genes (like epigenetic regulators: Lysinespecific histone demethylase and DNA methyltransferase 1), and this process is critical for the acquisition of CSC features[156].
Transcript-based expression analysis adds value to understanding of transcriptome in SC research
Measuring the expression of alternatively spliced mRNAs, instead of overall gene expression, is considered as a new and more accurate approach for marker discovery in cancer research[157]. For example, many apoptotic regulatory genes, such asBCL-x,encode for alternatively spliced protein variants with opposing functions: One apoptotic and the other anti-apoptotic[158]. In recent years, there has been a discernible shift in cancer diagnosis and therapy due to the perception about the role of genes and their proteins. Cancer can occur irrespective of changes in expression of a gene or protein, but rather as a result of aberrant splice variants that are linked to cancer progression and/or drug resistance and is compensated by the decreased expression of other splice variants originating from that same gene.
Future of AS
AS contributes to a range of phenotypic traits of tumours as they progress and metastasis, and is a potential target for gene therapy[6,9]. With the advent of nextgeneration sequencing technology (NGS) and bioinformatics approaches, studying AS patterns has been performed in both cancerous and non-cancerous cells with increasing details. However, assembled transcripts obtained from NGS technology are not complete because this technology could sequence short reads. Hence, NGS platform may not be suitable enough for AS analysis. To overcome this limitation, the Pacific Bioscience (PacBio) platform has been developed based on the single-molecule real-time sequencing technology. PacBio technology provides full-length transcript sequencing without assembly. The PacBio full-length transcriptome data is the most accurate source to investigate AS[159]. AS analysis has been performed in various plant and mammalian species using PacBio platform[160-162].
Some cancer-specific differential spliced isoforms have been identified which can be used in cancer diagnosis and as potential targets for selective anti-tumor treatments in the future[163]. Currently, single-cell transcriptomic studies have paved the way for analyzing the gene-level expression and identification of isoforms originating from the same gene, to determine the distinct gene expression signatures within cells, especially tumor cells. Also, single-cell techniques have provided a powerful method for identifying CSCs[164]. There are some controversies regarding the AS pattern in single-cells. Some evidence reported that several spliced transcripts exist in singlecells. Faigenbloomet al[165] measured pairs of included and skipped isoforms obtained from spliced exons in single-cells. While, other studies demonstrated that single-cells express only one transcript variant or the dominance of a single transcript variant[166,167]. Further work in single-cell AS analysis is required to unravel the potential future of using AS events to develop personalized medicine for various diseases including cancer.
CONCLUSION
Unravelling AS opens a new avenue towards the establishment of new diagnostic and prognostic markers of cancer progression and metastasis as well as the development of a new generation of anticancer therapeutics: Treatments that inhibit specific splice variants, rather than targeting genes. Although the significant roles of alternatively spliced transcripts in promoting self-renewal properties of CSCs have been identified,more studies are needed to identify the whole CSCs-related splicing events to strengthen the therapeutic benefits of AS in the future.
 World Journal of Stem Cells2021年10期
World Journal of Stem Cells2021年10期
- World Journal of Stem Cells的其它文章
- Translational products of adipose tissue-derived mesenchymal stem cells: Bench to bedside applications
- Unveiling the morphogenetic code: A new path at the intersection of physical energies and chemical signaling
- SOX transcription factors and glioma stem cells: Choosing between stemness and differentiation
- Retina stem cells, hopes and obstacles
- Considerations for the clinical use of stem cells in genitourinary regenerative medicine
- Age and genotype dependent erythropoietin protection in COVID-19