
Sin团簇/石墨烯(n≤6)结构稳定性和储锂性能的第一性原理计算*
2021-11-01 06:10沈丁刘耀汉唐树伟董伟孙闻王来贵杨绍斌
物理学报 2021年19期
沈丁 刘耀汉 唐树伟 董伟 孙闻 王来贵 杨绍斌†
1) (辽宁工程技术大学材料科学与工程学院,阜新 123000)
2) (辽宁工程技术大学力学与工程学院,阜新 123000)
目前,硅/碳复合材料是锂离子电池最有潜在应用前景的高容量负极材料之一,硅与碳材料的界面状态是影响其电化学性能的重要因素.本文在作为碳材料结构单元的石墨烯表面构建了Sin(n ≤ 6)团簇,采用基于密度泛函理论(DFT)的第一性原理方法研究了Sin 团簇/石墨烯(Sin/Gr)的几何构型、结构稳定性和电子性质.结果表明,当Si 原子数n ≤ 4 时,Sin 团簇优先以平行于石墨烯的二维构型沉积在石墨烯表面,当n ≥ 5时,Sin 团簇优先以三维立体构型沉积在石墨烯表面.随着n 的增大,Sin 团簇在石墨烯表面的热力学稳定性显著降低,两者之间的界面结合减弱,并且伴随着Sin 团簇与石墨烯之间的电荷转移也越来越少.同时还研究了Sin/Gr 复合构型的储锂能力,Li 原子主要存储在Sin 团簇临近的石墨烯表面和Sin 团簇周围,Sin 团簇与石墨烯复合形成的协同作用增强了Li 原子吸附的热力学稳定性.当n ≤ 4 时,存储2 个Li 原子有利于提高xLi-Sin/Gr 体系的热力学稳定性,继续增加Li 原子数x 会导致稳定性降低;当n ≥ 5 时,稳定性随着Li 原子数x 的增多而逐渐降低.
1 引言
锂离子电池具有容量高、循环寿命长等优点,在便携式电子设备领域得到广泛应用,并且在电动汽车领域展现了良好的发展势头.目前最常用的石墨负极储锂容量较低,仅为372 mA·h/g (LiC6),不能满足人们对高容量的需求.硅的储锂容量高(4200 mA·h/g),成为锂离子电池负极材料的研究热点[1].但是硅在储锂过程中体积膨胀大(超过300%),导致循环过程中容量衰减快,严重阻碍了硅在锂离子电池中的实际应用[2,3].
为了提高硅的循环性能,人们已经做出了巨大的努力,包括减小硅颗粒的尺寸[4,5]、形成多孔结构[6,7]、与其他金属合金化[8,9],或者与碳材料等形成复合结构[10−14].其中,将纳米尺度的硅颗粒与碳材料进行复合是目前最有效的途径.
Luo 等[15]以间苯二酚和甲醛为硬炭前驱体,采用原位聚合在纳米硅颗粒表面形成均匀包覆层,最后碳化形成核壳结构.他们发现碳壳厚度约为10 nm 的Si/C 复合材料表现出优异的电化学性能,在电流密度为500 mA/g 条件下首次可逆容量为2542 mA·h/g,库仑效率为74.9%,经过500次循环后容量仍然保留了1006 mA·h/g.Cai 等[16]采用SiCl4和Li 反应合成颗粒尺寸约为20 nm 的硅团聚体,然后再以乙炔为碳源进行气相沉积得到碳包覆的复合材料.该材料在电流密度为300 mA/g条件下,首次可逆容量为2242 mA·h/g,库仑效率达75.9%,比包覆前分别提高36.3%和67.9%,并且表现出优异的循环性能和倍率性能.林伟国等[17]以硅藻土为原料,采用镁热还原纳米多孔硅,再以沥青为软炭前驱体,与石墨混合后采用喷雾干燥和高温碳化,合成了纳米多孔硅/石墨/碳复合微球.该复合材料在电流密度为200 mA/g 条件下首次可逆容量为817 mA·h/g,库仑效率达84.0%,经过100 次循环后容量剩余765 mA·h/g.Zhu 等[18]以蔗糖、碳纳米管为碳源,采用喷雾干燥和高温碳化,合成了具有多级导电网络的硅/碳纳米管/碳复合材料,该材料具有显著的高倍率性能,在电流密度为7.5 A/g 条件下,可逆容量高达620 mA·h/g,同时首次库仑效率高达86.0%,经过100 次循环后容量保持率达80%.Yu 等[19]以还原氧化石墨烯(rGO)为碳源,该材料同样具有优异的倍率性能,在电流密度为0.2 A/g 条件下的可逆容量达2030 mA·h/g,在4 A/g 时经过200 次循环后容量保持率为99.2%,即使在8 A/g 时可逆容量高达1000 mA·h/g.
上述研究表明,碳材料一方面起到缓冲锂离子嵌入或脱出导致体积膨胀而产生的内应力,提高复合结构的结构稳定性和循环性能;另一方面还能够起到储锂作用,在改善循环性能的同时提供一定的储锂容量;另外碳材料还能提供电子传输通道,提高复合材料的导电性和倍率性能.因此硅与碳形成的复合材料成为众多理论和实验研究的热点.但是迄今为止硅与碳材料的界面结合情况以及相关理论研究非常缺乏,尤其是他们二者形成的复合材料的储锂机理研究更少.
采用基于密度泛函理论(DFT)的第一性原理计算可以从电子层面对复合材料的界面结合状态和储锂性能等进行预测,成为深入研究、选择和设计锂离子电池电极材料的一种重要方法[20−22].碳材料包括硬碳、软碳、碳纳米管、碳纤维和石墨烯等,种类繁多、结构复杂[23,24].为了避免不同构型的碳材料引起的巨额计算量,本文采用石墨烯作为碳材料的结构单元模型,在石墨烯表面构建Sin(n≤6)团簇.采用基于DFT 的第一性原理方法研究了石墨烯表面沉积Sin团簇的几何构型、结构稳定性和电子性质,同时还研究了它们的储锂能力.期望研究有助于从电子层面理解硅与碳材料的界面状态、相互作用以及储锂性能之间的关系.
2 理论计算方法
在作为碳材料结构单元的石墨烯表面,Si 原子逐步沉积形成Sin团簇.发生的反应可以描述为nSi+Gr↔Sin/Gr,其中Gr 为6×6 的石墨烯超胞,n为Si 原子数,Sin/Gr 为石墨烯表面沉积Sin团簇形成的结构.Gr 晶胞尺寸设置为14.76 Å×14.76 Å×20 Å,真空层设置为20 Å,足以避免周期性的影响.
采用基于DFT 的平面波赝势方法的CASTEP 软件包[25]对Sin/Gr 复合结构的几何构型、吸附能、差分密度电荷和态密度等性质进行理论计算.计算过程中交换关联能采用广义梯度近似(GGA)下的PBE 泛函[26]和范德瓦耳斯修正的Grimme 方法[27].采用超软赝势[28]来描述原子核与价电子之间的相互作用.通过收敛性测试,平面波展开的截断能设为500 eV,Monkhorst-Pack 型k点网格分布[29]设为5×5×1,原子的总能量收敛标准设为1.0×10–5eV/atom,力收敛标准设为0.02 eV/Å.
为了研究石墨烯表面沉积Sin团簇形成复合结构的热力学稳定性,定义Si 原子的平均吸附能ΔEab为
为了研究Li 原子在Sin/Gr 复合结构的热力学稳定性,定义Li 原子的平均吸附能ΔEab为
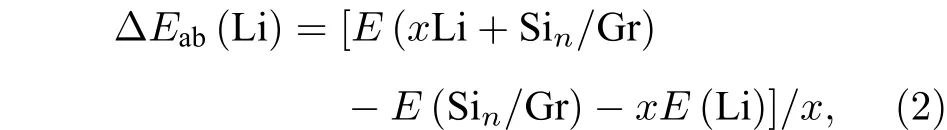
(1)式和(2)式中E(xLi+Sin/Gr),E(Sin/Gr),E(Gr),E(Sin)和E(Li)分别是优化的xLi-Sin/Gr 体系、Sin/Gr 复合构型、6×6 的石墨烯超胞的总能量、Sin团簇和Li 原子的能量,n和x分别是Si 原子和Li 原子的数量.当ΔEab为负值,表明Sin团簇能够稳定沉积在石墨烯表面,或Li 原子能够稳定吸附在Sin/Gr 复合构型.
3 结果与讨论
3.1 Sin/Gr 结构稳定性和电子结构
单个原子在石墨烯表面的沉积有三个高对称性位置,即空位(H 位)、C 原子上方(T 位)、桥位(B 位),Si 原子在这三个位置的沉积状态如图1 所示.由图1(a)可知,经过几何优化后,Si 原子在石墨烯B 位的吸附能量ΔEab最低,为–1.216 eV;Si原子在T 位和H 位的ΔEab依次增大,分别为–1.145和–0.738 eV.计算结果表明,单个Si 原子优先沉积在石墨烯B 位,T 位次之,H 位最不稳定.这与Gao 等[30]和Aktürk 等[31]的研究结果一致,他们认为Si 原子在石墨烯B 位或T 位形成较强的化学吸附,而在H 位形成以范德瓦耳斯力为主的物理吸附.由图1(b)可知,当Si 原子在石墨烯B 位和T 位时形成的Si—C 键长dSi—C分别为2.093和2.053 Å,都比碳化硅晶体中Si—C 键的键长(1.89 Å)[32]长.Si 原子与石墨烯发生相互作用,导致石墨烯碳平面发生上下位移,当Si 原子在石墨烯B 时位移高度Δh值达最大值,为0.052 Å,这是由于石墨烯吸附Si 原子以后,通过调整C 原子的位置,使体系能量达到最低值,形成最稳定的吸附结构.
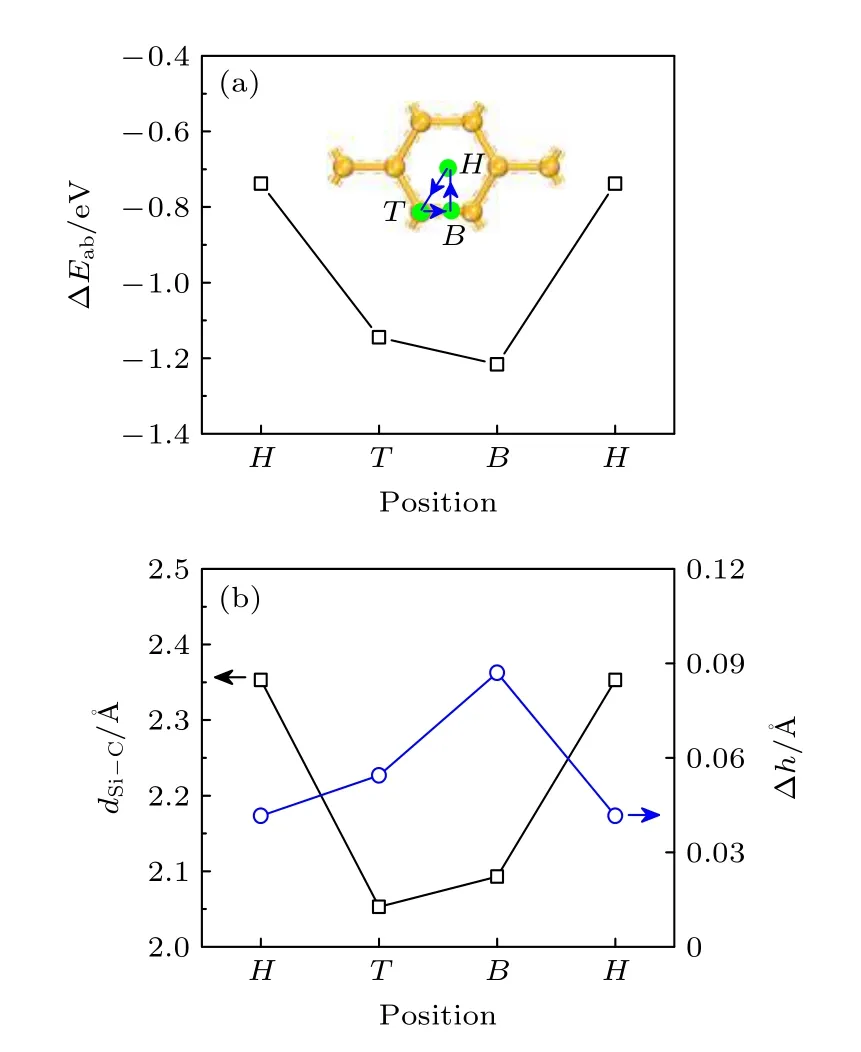
图1 单个Si 原子在石墨烯表面的吸附行为 (a)吸附能量ΔEab;(b) Si—C 键长dSi—C 和C 原子位移高度ΔhFig.1.Adsorption behavior for a single Si atom on graphene:(a) Adsorption energy ΔEab;(b) length of Si—C dSi—C and shift height Δh of C atom.
当石墨烯表面沉积超过2 个Si 原子时,Sin团簇沉积在石墨烯表面的构型如图2 所示,其吸附能和结构参数列于表1.由图2 可知,当n≤ 4 时,Sin团簇似乎都希望以平行于石墨烯平面的二维构型沉积在石墨烯表面.稳态的Si2二聚体中2 个Si 原子优先沉积在石墨烯间隔的T 位,Si 原子平均沉积高度h为2.532 Å.Si2中Si 原子与C 原子键合,形成Si—C 键的平均键长dSi—C为2.223 Å.Si2中Si—Si 键长dSi—Si为2.262 Å,比真空中Si2二聚体的键长(2.290 Å)缩短.稳态的Si3团簇以等腰三角形沉积在石墨烯表面,其中1 个Si 原子沉积在石墨烯T 位,2 个Si 原子沉积在石墨烯B 位.Si3中Si 原子平均沉积高度h为3.365 Å,比Si2显著增高.Si3中Si 与C 原子形成Si—C 键的平均键长dSi—C也显著增长,为3.456 Å.Si3中Si—Si 键平均键长dSi—Si为2.187 Å.稳态的Si4团簇以平行四边形沉积在石墨烯表面,4 个Si 原子沉积在石墨烯T 位,平均沉积高度h为3.235 Å.Si4团簇中Si—C 键平均键长dSi—C为3.246 Å,Si—Si 键平均键长dSi—Si为2.335 Å.
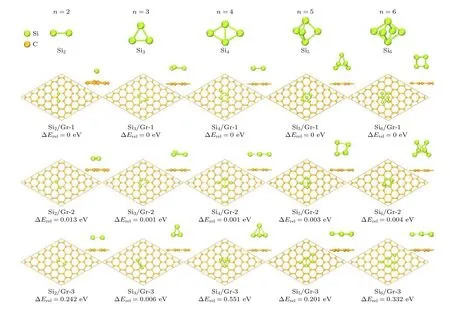
图2 石墨烯表面Sin 团簇的构型 (ΔErel 为亚稳态与稳态构型的总能量之差)Fig.2.Configuration for Sin clusters on graphene (ΔErel is the difference of total energy between metastable and steady-state configurations).
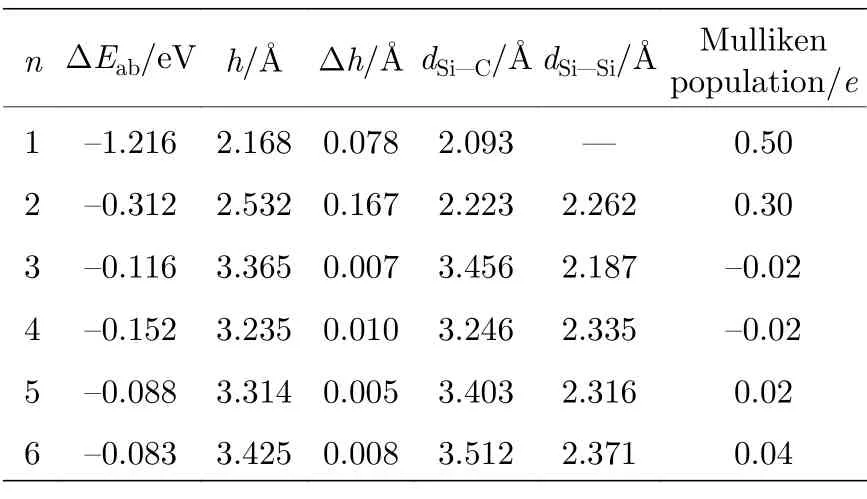
表1 石墨烯表面Sin 团簇的平均吸附能、结构参数和Mulliken 布局Table 1.Average adsorption energy,structural parameters and Mulliken population for Sin clusters on graphene.
当n≥ 5 时,Sin团簇通常以三维立体构型沉积在石墨烯表面.稳态的Si5团簇以保持真空态的双三棱锥立体构型沉积在石墨烯表面,临近石墨烯底层中的2 个Si 原子沉积在石墨烯B 位附近,第二层中的2 个Si 原子位于石墨烯T 位,第三层中的Si 原子位于石墨烯H 位.Si5团簇中底层Si 原子平均沉积高度h为3.314 Å,Si—C 键的平均键长dSi—C为3.403 Å,Si—Si 键的平均键长dSi—Si为2.316 Å.稳态的Si6团簇也以保持真空态的双四棱锥立体构型沉积在石墨烯表面,临近石墨烯底层的3 个Si 原子形成的Si—Si—Si 面沉积在石墨烯表面B 位附近,第2 层中的3 个Si 原子沉积在石墨烯表面H 位.Si6团簇中底层的Si 原子平均沉积高度h为3.425 Å,Si—C 键平均键长dSi—C为3.512 Å,Si—Si 键平均键长dSi—Si为2.371 Å.
由表1 可知,随着Si 原子数n的增多,Sin团簇在石墨烯表面的平均吸附能ΔEab整体呈现增大并接近0 的规律,沉积高度h整体呈现逐渐升高的规律.尤其是当n≥ 3 时,吸附能量ΔEab和沉积高度h大幅度增大,这表明Sin团簇在石墨烯表面的热力学稳定性显著降低.这从Mulliken 电荷转移也能看出类似规律,Si2二聚体向石墨烯转移的电荷量为0.30e,当n≥ 3 时,Sin团簇向石墨烯转移的电荷量可以忽略不计.同时这也体现在石墨烯中C 原子在垂直于石墨烯方向发生位移(Δh)的变化规律上,Si2中Si 原子与石墨烯的C 原子发生较强的相互作用,导致C 原子发生位移显著,Δh值达0.169 Å;当n≥ 3 时,Sin团簇对石墨烯的相互作用较弱,Δh值仅为0.005—0.010 Å.
为了进一步了解Sin团簇与石墨烯之间的相互作用,图3 对比研究了孤立的Sin团簇和Sin/Gr复合材料的总态密度和分波态密度.对于单个Si 原子和Sin团簇,Si 原子表现出窄而尖锐的能带,其特征是一组离散的能级,如图3(a)所示.当Si 原子或Sin团簇与石墨烯复合后,Si 原子或Sin团簇的分波态密度呈现离域加宽的能带,这在单个Si 原子和Si2二聚体中表现极为显著,如图3(b)所示;同时导致石墨烯在费米能级附近出现微弱的小峰,如图3(c)所示,这是由Si 原子的p 电子和石墨烯中C 的p 电子发生明显的相互作用所致.随着Si 原子数量的增多,Sin团簇与石墨烯之间的相互作用较弱.当n≥ 4 时,复合结构中的Sin团簇的态密度又呈现出窄而尖锐的能带,费米能级附近的态密度为零;同时石墨烯的态密度与复合前相比变化不明显.由图3(d)可知,石墨烯呈现半金属性,在费米能级上的态密度为零.当Si 原子或Sin团簇与石墨烯复合后,Si 原子的p 电子与石墨烯的p 电子轨道穿越费米能级,在费米能级出现态密度,导致复合结构的电子导电性和金属性增强.但随着Si 原子数的增多,当n≥ 4 时,复合结构在费米能级上的态密度又逐渐降低至零,电子导电性降低.
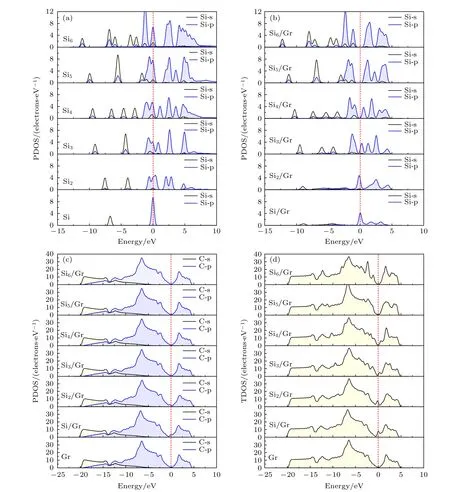
图3 孤立Sin 团簇和Sin/Gr 复合材料的态密度图 (a) 孤 立Sin 团簇的分波态密度;(b),(c) Sin/Gr 复合材 料的分 波态密 度;(d) Sin/Gr 复合材料的总态密度Fig.3.Density of states for isolated Sin clusters and Sin/Gr composites:(a) Partial density of states for isolated Sin clusters;(b),(c) partial density of states for Sin/Gr composites;(d) total density of states for Sin/Gr composites.
3.2 Sin/Gr 构型的储锂性能和电子结构
石墨烯和Sin团簇都具有较高的储锂能力,为了研究二者复合形成Sin/Gr 结构的储锂能力,将临近Sin团簇的石墨烯表面、Sin团簇的侧面以及顶部设置为Li 原子的初始吸附位置,经过结构优化后得到Sin/Gr 复合结构吸附不同Li 原子数的稳态构型(xLi-Sin/Gr),如图4 所示.由图4 可知,Li 原子主要存储在Sin团簇临近的石墨烯表面,随着Li 原子数的增多,Li 原子逐渐存储在Sin团簇周围;并且吸附的Li 原子会导致部分Sin团簇的构型或在石墨烯的位置发生改变.
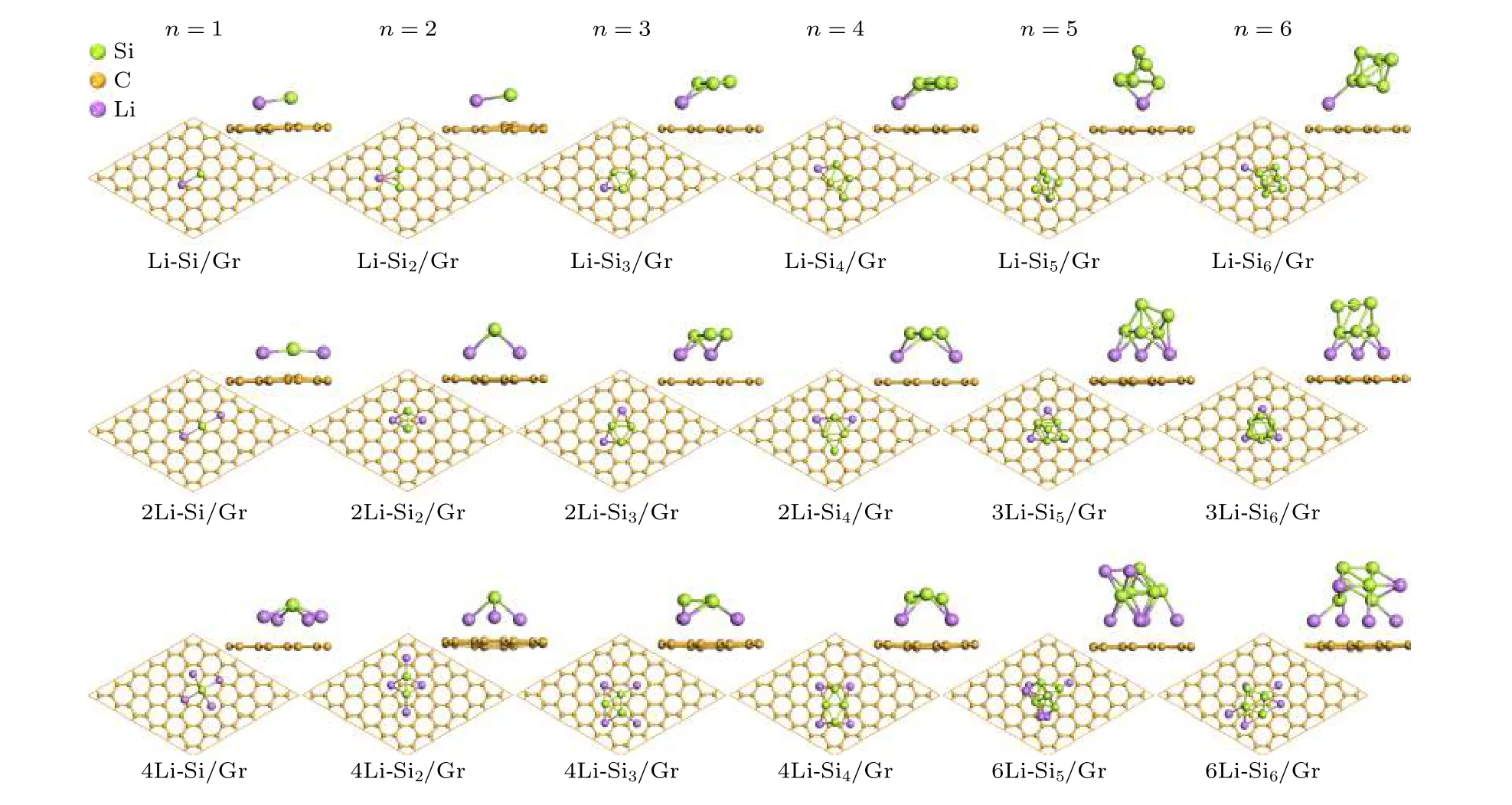
图4 Sin/Gr 复合结构吸附不同Li 原子数的稳态构型Fig.4.Configuration for Sin/Gr system with different Li atom numbers.
图5 为稳态构型(xLi-Sin/Gr)中Li 原子数x与Li 原子平均吸附能ΔEab之间的关系.可以看出,Sin/Gr 复合结构的Li 原子平均吸附能ΔEab与石墨烯相比均明显较低,表明Sin团簇与石墨烯复合形成的协同作用增强了Li 原子吸附的热力学稳定性.当n≤ 4 时,Sin/Gr 复合结构的ΔEab值随着Li 原子数x的增多均呈现先降低后增加的趋势,但变化幅度不明显;这是由于Li 原子主要吸附在二维Sin团簇临近的石墨烯表面,当存储2 个Li 原子时有利于提高结构的热力学稳定性,但更多的Li 原子容易导致结构稳定性降低.对于n≥ 5时,Sin/Gr 复合结构的ΔEab值随着x的增多呈现增大的趋势,这表明三维Sin团簇与石墨烯形成复合构型的结构稳定性随着Li 原子数的增多而逐渐降低.
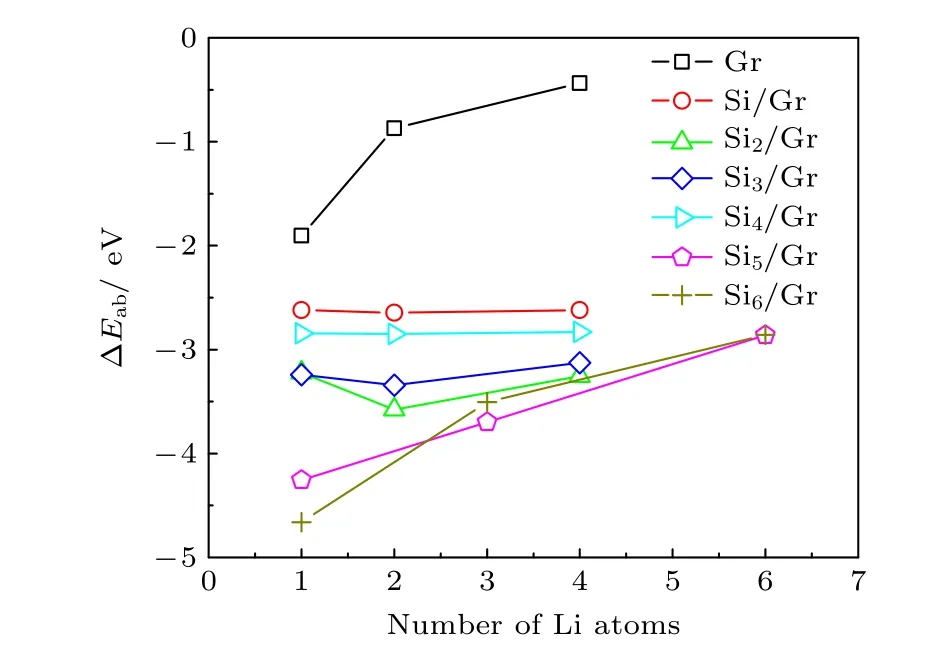
图5 稳态构型(xLi-Sin/Gr)中Li 原子数与平均吸附能ΔEab 的关系Fig.5.Relationship between the average adsorption energy ΔEab and the number of Li atoms in the steady-state configuration (xLi-Sin/Gr).
由于石墨烯表面Si5团簇模型具有较高的对称性,选择Si5/Gr 复合结构研究储存Li 原子过程中的电荷转移和成键情况,对xLi-Si5/Gr 体系(x=1,3 和6)的稳态构型进行电子结构分析,结果如图6 所示,其中电子密度差分析图中红色区域为失电子区域,蓝色区域为得电子区域.由图6 可知,当Si5/Gr 体系吸附Li 原子后,Li 原子周围临近石墨烯附近出现明显的蓝色得电子区域和红色失电子区域,表明Li 原子与石墨烯之间发生明显的电子转移,Li 原子与C 原子形成的Li—C 键具有一定的离子键属性.
Mulliken 布局计算结果表明,当Si5/Gr 体系吸附1 个Li 原子,Li 原子的部分电子转移至石墨烯(0.36e),部分电子转移至Si5团簇(0.74e).当Si5/Gr 体系吸附3 个Li 原子,Li 原子分别向石墨烯和Si5团簇转移1.51e和1.78e电荷.当Si5/Gr体系吸附6 个Li 原子,Li 原子分别向石墨烯和Si5团簇转移2.60e和3.54e电荷.表明随着Li 原子数x增多,xLi-Si5/Gr 体系中Li 原子向石墨烯和Si5团簇转移的电荷量增大.
电子局域密度函数(ELF)[33,34]可以对电子进行定域分析,值域为0—1.当ELF=1 时,电子高度局域化,表现为最强的共价键;当ELF=0.5 时,表现为金属键;当0 ≤ ELF <0.5 时显示为较强的离子键.由图6 可知,xLi-Si5/Gr 体系中,石墨烯内的C—C 键和Si5团簇内的Si—Si 键都主要为共价键,Li 原子与C,Si 原子形成的Li—C 键和Li—Si键主要为含有一定共价属性的离子键.
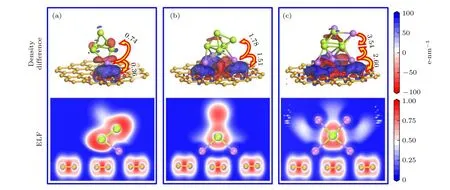
图6 xLi-Si5/Gr 体系(x=1,3 和6)稳态构型的电子结构分析(图中数据为Mulliken 布局)Fig.6.Electronic structure analysis of steady-state configuration of xLi-Si5/Gr system (x=1,3 and 6) (The data in figure is Mulliken population).
4 结论
本文采用基于DFT 的第一性原理方法,研究了石墨烯表面沉积Sin团簇(n≤ 6)的几何构型、结构稳定性和电子性质.当Si 原子数n≤ 4 时,Sin团簇优先以平行石墨烯的二维构型沉积在石墨烯表面;当n≥ 5 时,Sin团簇优先以三维立体构型沉积在石墨烯表面.随着Si 原子数的增多,Sin团簇在石墨烯表面的热力学稳定性显著降低,两者之间的界面结合减弱,Sin团簇与石墨烯之间的电荷转移也越来越少.同时还进一步计算了Sin/Gr复合结构中Li 原子的吸附能,以研究Sin/Gr 复合结构的储Li 能力.Li 原子主要存储在Sin团簇临近的石墨烯表面和Sin团簇周围,Sin团簇与石墨烯复合形成的协同作用增强了Li 原子吸附的热力学稳定性.当n≤ 4 时,存储2 个Li 原子有利于提高xLi-Sin/Gr 体系的热力学稳定性,继续增加Li 原子数会导致稳定性降低;当n≥ 5 时,稳定性随着Li 原子数的增多而逐渐降低.研究结果期望有助于从电子层面理解硅与碳材料的复合界面状态,以及作为锂离子电池电极材料的储锂性能.
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
桂林电子科技大学学报(2020年1期)2020-12-18
岩石矿物学杂志(2020年1期)2020-06-05
湖南工程学院学报(自然科学版)(2020年1期)2020-03-26
青岛大学学报(工程技术版)(2019年2期)2019-09-10
北京航空航天大学学报(2017年10期)2017-04-20
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
中学化学(2015年5期)2015-07-13
