
UV工艺降解阿特拉津机理及对DBPFP的影响规律
2021-10-26 13:29刘玉灿王玉霞朱玉良纪现国段晋明
中国环境科学 2021年10期
刘玉灿,王 颖,王玉霞,朱玉良,纪现国,张 岩,段晋明,李 伟
UV工艺降解阿特拉津机理及对DBPFP的影响规律
刘玉灿1*,王 颖1,王玉霞2,朱玉良1,纪现国1,张 岩1,段晋明3,李 伟3
(1.烟台大学土木工程学院,山东 烟台 264005;2.华北水利水电大学环境与市政工程学院,河南 郑州 450046;3.西安建筑科技大学环境与市政工程学院陕西 西安 710055)
对不同UV光氧化工艺降解水溶液中阿特拉津(ATZ)的动力学和机理,以及对后续氯化处理过程中溶液需氯量和消毒副产物生成势(DBPFP)的影响规律与机理进行了系统研究.结果表明,ATZ在不同UV光氧化工艺中的降解均符合准一级反应动力学.ATZ在单独UV辐照工艺中的去除效率相对较低;UV/H2O2工艺对ATZ具有相对较高的去除效率,且其去除率随H2O2浓度的增大呈现出先增加后降低的变化趋势;UV/TiO2工艺降解去除ATZ的效率较单独UV辐照和UV/H2O2工艺低,ATZ在UV/TiO2工艺中的降解与溶液透光率和氧化活性物种(ROS)生成量存在直接关系;UV/H2O2/TiO2工艺中,ATZ的降解速率较UV/TiO2工艺有所提高.ATZ水溶液经不同UV光氧化工艺预处理和氯化处理后,均检出了5种消毒副产物(DBPs),其中三氯甲烷(TCM)和三氯丙酮(TCP)为主要氯化DBPs.本研究表明,在不同UV光氧化预处理过程中,ATZ具有不同的降解路径,进而对ATZ水溶液在后续氯化过程中的DBPPF产生显著影响.
阿特拉津;高级氧化工艺;氯化消毒副产物;降解机理
阿特拉津(ATZ)是一种选择性内吸传导型除草剂,具有较高的水溶性(25ºC时的溶解度为33mg/L),残留在土壤中的ATZ可通过下渗和地表径流进入水体,从而对天然水体造成污染[1].由于ATZ的大量使用且其具有难降解特性,导致ATZ在天然水体中的检出率及浓度均相对较高.
ATZ在欧盟于2003年禁用,但在美国、中国和其他国家仍被广泛使用,故致天然水体不同程度地受到了ATZ的污染.例如,美国自2003年以来启动了“阿特拉津监测计划”,其中宾夕法尼亚州奇利斯夸克溪中ATZ的检出率高达93.3%[2];ATZ是中国最常用的除草剂之一,年使用量超过10000t[3],长江流域重庆段水体中ATZ的浓度为5.6~25.6ng/L,平均浓度为10.9ng/L[4].有研究表明,ATZ对哺乳动物具有内分泌干扰、生殖异常、致癌性等副作用,且其在天然水体中能够持续几十年乃至更长时间,故其成为了影响人类健康的常见污染物之一[5].由于ATZ对生态环境、农业生产及人体均具有较大危害,因此已被美国环保署和中国生态环境部列入水体优先控制污染物清单之中.
常规给水处理工艺对有机农药的去除效率均相对较低,高级氧化工艺(AOPs)因具有运行稳定、降解速度快、去除效率高等优点,受到越来越多的关注[6].UV光催化工艺是典型的AOPs,其产生的羟基自由基(•OH)的氧化还原电位高达2.8V,能将大多数有机污染物无选择性地矿化去除[7].
由于目前尚无统一规律可用来解释不同UV光氧化工艺中ATZ的降解途径及对后续溶液需氯量和氯化消毒副产物生成势(DBPFP)影响规律与机理的报道.因此,本文拟对不同UV光氧化工艺中ATZ的降解动力学和产物生成种类进行系统地研究,揭示UV光氧化预处理对ATZ溶液在后续氯化处理阶段中DBPFP的影响规律与机理,以期能从另一个视角评估UV光氧化处理在去除饮用水中ATZ的适用性,进而为饮用水处理中UV光氧化预处理的选用与优化提供理论依据和技术支持.
1 材料与方法
1.1 材料
ATZ(纯度>98.8%)购买于Sigma–Aldrich公司.二氧化钛(TiO2,P25,80%锐钛矿和20%金红石)购买于Evonik Degussa公司.甲基叔丁基醚(MTBE,GC级)、乙腈(HPLC级)和丙酮(GC级)购买于Merck公司.氢氧化钠(GR)、硫酸(GR)、过氧化氢(30%/)和次氯酸钠(5%有效氯)购买于国药集团化学试剂有限公司.磷酸二氢钠(GR)和磷酸氢二钠(GR)购买于科密欧化学试剂有限公司.超纯水(18.2MΩ·cm, TOC<1μg/L)由实验室配置的Purelab Ultra Analytic系统制取(ELGA,UK).
消毒副产物(DBPs)标准溶液均购买于Sigma– Aldrich公司.包括:(1)HAAs混标(2000μg/mL),含一氯乙酸(MCAA)、二氯乙酸(DCAA)、三氯乙酸(TCAA)、一溴乙酸(MBAA)、二溴乙酸(DBAA)、三溴乙酸(TBAA)、溴氯乙酸(BCAA)、一溴二氯乙酸(BDCAA)和二溴一氯乙酸(DBCAA);(2)EPA 551B混标(2000μg/mL),含二氯乙腈(DCAN)、溴氯乙腈(BCAN)、二溴乙腈(DBAN)、三氯乙腈(TCAN)、三氯硝基甲烷(TCNM)、1,1–二氯丙酮(1,1–DCP)和1,1,1–三氯丙酮(1,1,1–TCP); (3)THMs混标,含三氯甲烷(TCM)、一溴二氯甲烷(BDCM)、二溴一氯甲烷(DBCM)和三溴甲烷(TBM);(4)一氯乙腈(MCAN,纯度>99.0%)、一溴乙腈(MBAN,纯度>98.0%)和1,2–二溴丙烷(内标IS,纯度>99.0%).
1.2 光化学实验
天然水体及实际水处理中水的pH值多近中性,因此本研究以低压汞灯为光源,使用2mmol/L的磷酸缓冲盐将ATZ(5mg/L)水溶液的pH值调至7.0,并在光照前向ATZ溶液中分别加入5mg/L或30mg/L的H2O2或/和TiO2.光化学反应器型号为BL–GHX– V(上海比朗仪器有限公司),实验程序根据文献[8]并结合本研究实际情况确定.

图1 低压汞灯发射光谱
使用乙腈配制浓度为2.5g/L的ATZ储备液,然后使用超纯水(UPW)将其稀释成5mg/L的ATZ水溶液.采用UV/H2O2工艺时,先于光照前向配制好的ATZ溶液中加入一定浓度H2O2.采用UV/TiO2工艺时,首先将准确称量的TiO2粉末加入到UPW中,用玻璃棒搅拌后超声30min,以便TiO2完全分散,后加入ATZ标准溶液和缓冲盐定容至250mL.反应过程中间隔取样,并及时测定分析,否则将其置于4ºC冰箱中避光保存,且不超过一周.
1.3 氯化实验
按照APHA标准方法5710要求,对UV光氧化后的ATZ水溶液进行氯化实验.首先使用1mol/L的H2SO4或NaOH将ATZ溶液的pH值调至7.0,之后加入一定量的NaClO,保证反应24h后的自由性余氯浓度维持在(1.0±0.5)mg/L范围内,pH值处于7.0±0.2范围内.氯化处理时,向样品中加入NaClO后,快速搅拌15s,并立即将50mL样品转移至棕色玻璃容量瓶中,40mL样品转移至带聚四氟乙烯衬垫螺纹帽的萃取瓶中顶空保存.加氯后的样品均置于20ºC恒温培养箱中避光保存,反应24h后,对每个样品的自由性余氯进行测定(余氯浓度不满足要求的样品舍弃重新进行试验),然后向样品中加入过量硫代硫酸钠,阻止氯化反应的进行.
本研究中,自由氯浓度采用DPD比色法[9]测定,H2O2浓度采用碘量法测定[10].10种卤乙酸使用超高效液相色谱–电喷雾离子源–三重四极杆质谱联用仪(UPLC–ESI–MS/MS)检测,ACQUITY™ UPLC HSS T3 色谱柱 (2.1mm×50mm×1.8μm,Waters,USA)用于色谱分离.ATZ及其产物的测定分析使用UPLC–ESI–MS/MS,ACQUITY™ UPLC BEH C8色谱柱(2.1mm×100mm×1.7μm,Waters,USA)用于色谱分离.全扫模式下采用ESI+和ESI–模式同时采集,/范围设为50~500,采集时间为0~28min,扫描速度为0.2secs/scan.为获得降解产物的更多结构信息,在子离子模式下对其进行了诱导碰撞解离,并对碰撞能量(15~35eV)进行优化.
色谱条件:流动相由甲醇(A)和超纯水(B)组成,流速为0.2mL/min.洗脱梯度为:10%A保持3min,随后在15min内升至70%A;然后在4min内升至100%A,并保持3min;后降至10%A,并保持3min(总时间为28min).进样量为10μL,色谱柱和样品室的温度分别设为35ºC和25ºC.
质谱条件:采用ESI源(ESI+、ESI–),毛细管电压和锥孔电压分别设为3.3kV和35V;离子源和脱溶剂气的温度分别为110ºC和350ºC;脱溶剂气(N2)为500L/h,锥孔气(N2)流量为30L/h,子离子模式下碰撞气(Ar)的流量为0.12mL/min.
13种挥发性DBPs使用气相色谱–电子轰击源–三重四极杆质谱联用仪(GC–EI–MS/MS)检测. MS/MS作为检测仪器,Mass Hunter B.04.00用于仪器控制、实时显示和数据采集.进样口采用不分流模式,进样体积为1.0µL,HP–5MS色谱柱(5%二苯基–95%二甲基聚硅氧烷,30m×0.25mm×0.25μm, Agilent)用于色谱分离.离子源为电子轰击源(EI),N2作为碰撞气,流速为1.5mL/min,淬火气(N2)流量为2.25mL/min;离子源和四极杆的温度分别设为230oC和150oC;灯丝发射电流为35µA,溶剂延迟时间为2.5min;所有扫描均在高分辨率条件下运行,每个扫描段的驻留时间为80~240ms.
2 结果与讨论
2.1 反应条件对ATZ降解动力学的影响
暗对照实验中,ATZ在120min内的浓度降低均小于2%,表明ATZ能在水溶液及含H2O2或/和TiO2的水溶液中稳定存在.因此,pH=7.0条件下的光照试验中,ATZ浓度的降低来自光解和光催化氧化降解.本研究中,ATZ(5mg/L)的降解遵循准一级反应动力学[11],ATZATZ0、ln(ATZATZ0)与光照时间的关系见图2(5mg/L H2O2或/和TiO2)和图3(30mg/L H2O2或/和TiO2).其中,ATZ的动力学见方程(1).
dATZ/d=–·ATZ·(1)
ATZ浓度与UV时间的半对数关系见方程(2).
ln(ATZ/ATZ0)=–=–=–obs(2)
式中:ATZ0和ATZ分别为初始时间和时间时的ATZ浓度,mg/L;为光射时间,min;为光通量率, mW/cm2;为光通量,mJ/cm2,等于×;为反应速率常数,cm2/mJ;obs为反应速率常数,1/min,等于·.
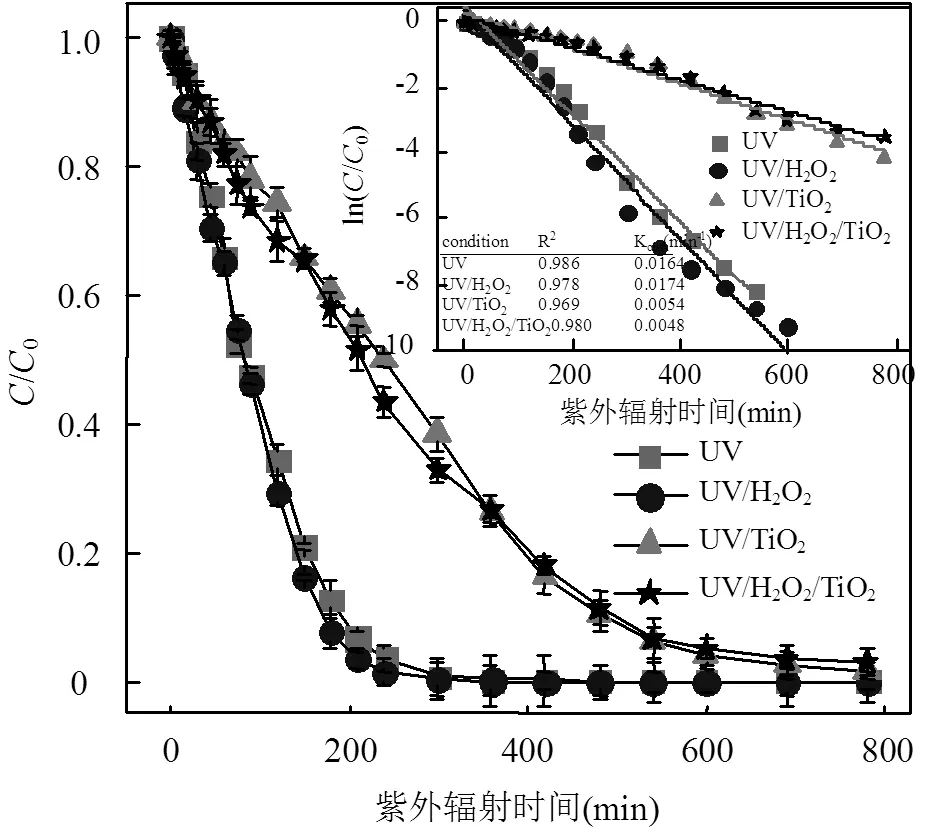
图2 UV光氧化工艺对ATZ降解动力学的影响(5mg/LH2O2或/和TiO2)
由图2可知,不同UV光氧化工艺中,光照初期ATZ的浓度降低的速率均较快,但随着ATZ浓度的降低,其降解速率逐渐降低.其中,单独UV辐照工艺对ATZ的去除速率相对较大[12],H2O2(5mg/L)的加入能够提高ATZ的降解速率,而TiO2(5mg/L)的加入却显著降低ATZ的降解速率,TiO2和H2O2同时加入时ATZ的降解速率较低于其在单独UV辐照工艺、UV/H2O2(5mg/L)和UV/TiO2(5mg/L)工艺中的降解速率.
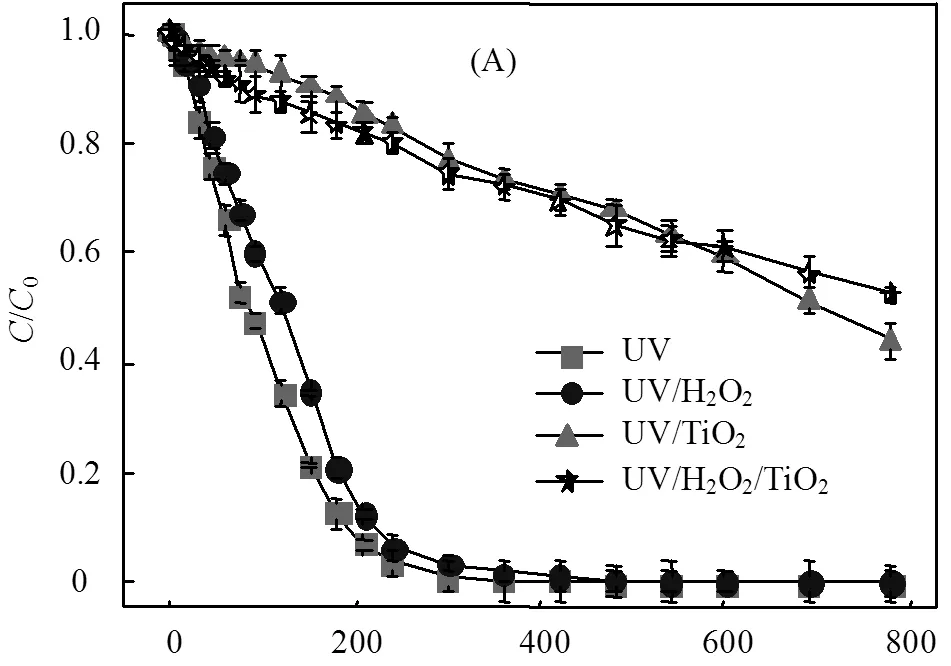
由图3(A)可知,单独UV辐照工艺中ATZ的降解速率较UV/H2O2(30mg/L)工艺快,这表现出了与图2相反的结果.UV/TiO2(30mg/L)和UV/H2O2/TiO2(30mg/L)工艺中,ATZ的降解速率均较慢,且在光照处理780min内不能将ATZ完全去除.H2O2或/和TiO2(30mg/L)的加入均未提高ATZ的降解速率,见图3(B).
2.2 不同工艺中ATZ的降解产物生成及机理
使用UPLC–ESI–MS/MS对经不同UV光氧化工艺处理前后的ATZ溶液进行了测定分析,对比ATZ经不同UV光氧化工艺处理前后的提取离子色谱图(EIC),确定了不同UV光氧化工艺中ATZ的主要产物峰,见图4
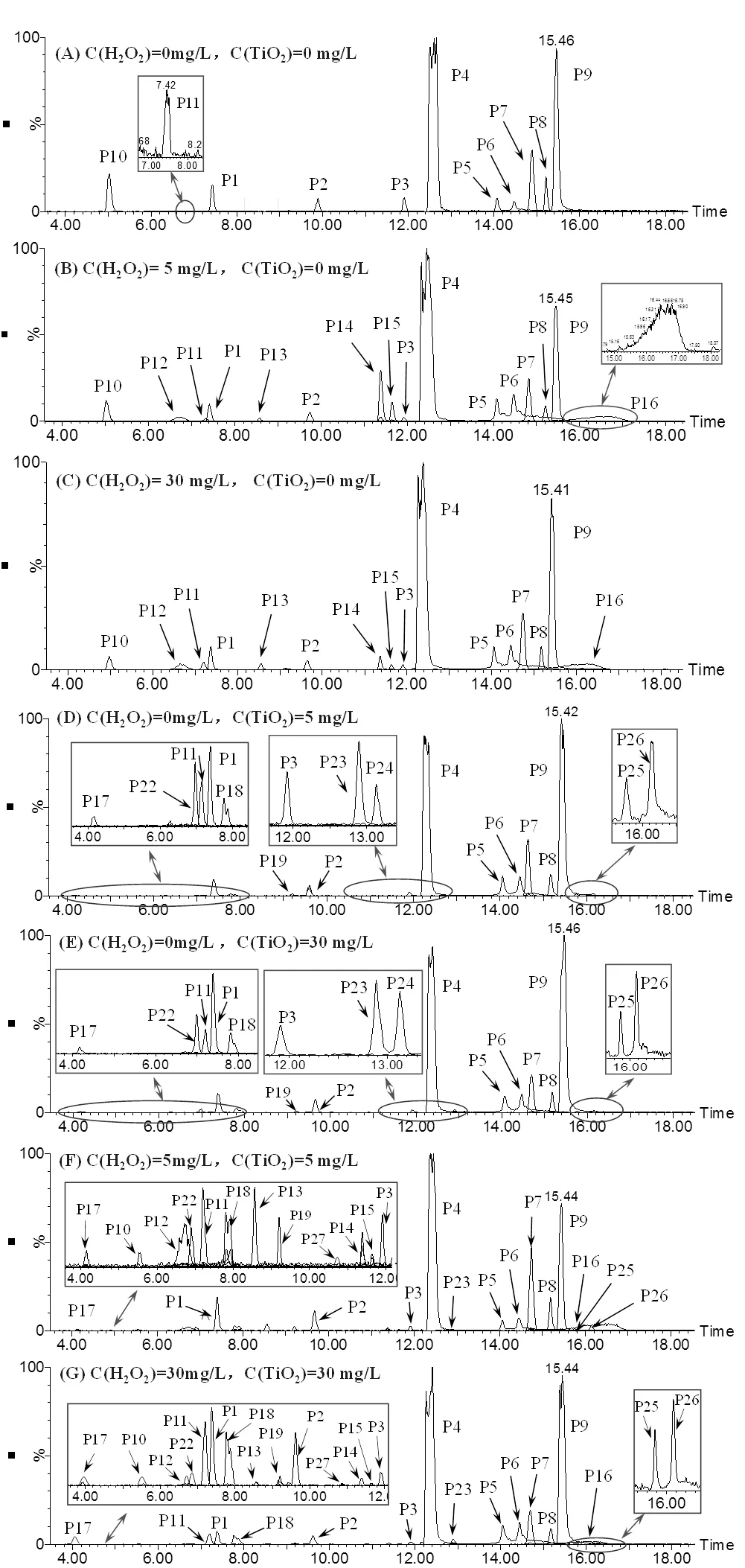
图4 ATZ溶液经不同UV光氧化工艺处理后的提取离子色谱
2.2.1 单独UV辐照工艺中ATZ产物生成及机理 ATZ的光解过程包括直接光解和间接光解,但这些过程的速率通常较低.为研究ATZ在UV辐照工艺中氧化产物的生成规律,本研究对初始浓度为5mg/L的ATZ水溶液进行了UV光照处理.经单独UV辐照工艺处理后,ATZ及其10种降解产物被检出[图4(A)].为便于讨论分析,将图4(A)中的色谱峰分别标记为P1、P2、P3、P4、P5、P6、P7、P8、P9、P10和P11,其中P9为目标物ATZ[13].基于测得的色谱、质谱信息,并结合文献[14]推导出了P1~P11可能对应物质的分子结构(表1).
UV光照处理时,目标物会首先分解成中间产物,部分产物可进一步分解为CO2、H2O等无机物,但该过程的反应速率通常较低.有研究表明,水分子在波长小于190nm的UV照射下才会产生•OH和水合电子,故UV–254nm光照作用下,ATZ浓度的降低均为ATZ吸收光量子(hν)后发生的直接光解所致.综上所述,ATZ浓度的降低均为UV照射作用下的缓慢光解,而非•OH氧化所致.ATZ在单独UV辐照工艺中发生了10种主要反应类型,相关反应类型见文献[8].
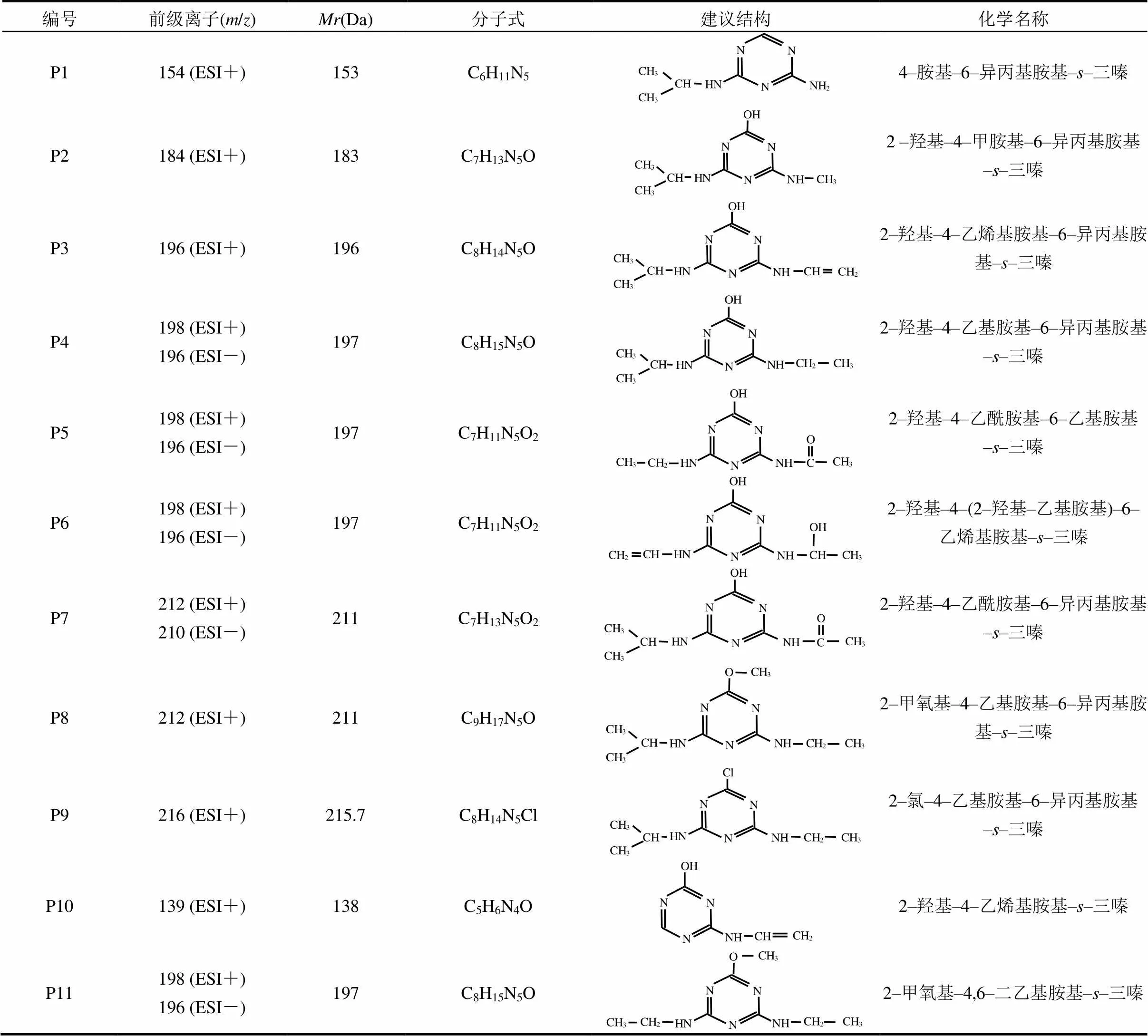
表1 UV辐照工艺中阿特拉津及其降解产物的建议分子结构式及相关信息
2.2.2 UV/H2O2工艺中ATZ产物生成及机理 UV/H2O2工艺具有较高的污染物去除效率和有机物矿化度,且该工艺简单易行,故被广泛用于水和废水中微污染物或难降解有机物的深度处理.本研究对H2O2加入量分别为5mg/L和30mg/L的ATZ水溶液进行了处理,考察了ATZ在UV/H2O2工艺中氧化产物的生成规律.结果表明,UV/H2O2(5mg/L)工艺中[图4(B)],除检出了P1~P11[15]外,亦检出了新产物,将其分别标记为P12、P13、P14、P15和P16(表2).由图4(B)和(C)可知,H2O2浓度的增加未改变色谱峰的数量和位置,即ATZ降解产物的种类未发生改变.
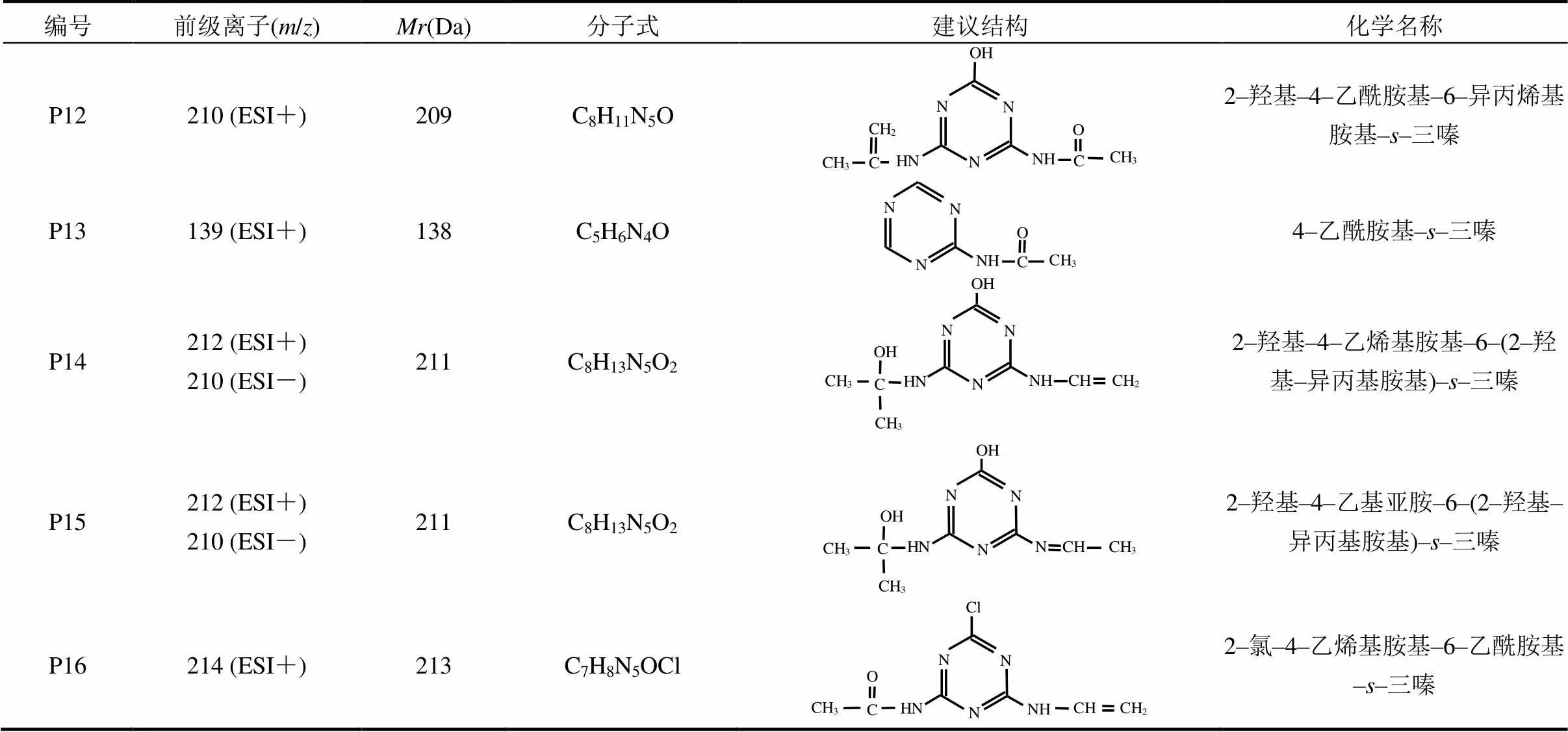
表2 UV/H2O2工艺中阿特拉津降解产物的建议分子结构式及相关信息
UV照射条件下,H2O2浓度由0增至5mg/L时,ATZ的去除率显著增加[16],这归因于H2O2光解产生了•OH[式(3)],•OH可优先与ATZ的疏水基团反应,从而产生亲水性产物,且H2O2的加入可致三嗪结构开环,碳结构的裂解及开环可产生更多的反应位点.H2O2浓度的增加可提高体系中•OH的含量.这是由于H2O2可在UV光照作用下发生均裂,促使体系中生成更多的•OH[式(3)],而•OH是光催化体系中的主要活性氧化物种(ROS),故能使反应体系具有更强的氧化能力.因此,在UV光解和•OH氧化[式(4),式(5)]的双重作用下,ATZ能够具有更快速的氧化降解速率,但当反应体系中H2O2浓度过高时,ATZ的降解速率反而出现了降低.这是由于H2O2会与ATZ分子竞争hν,从而造成ATZ分子吸收的hν总量减少,过量H2O2还会与•OH发生反应,生成活性相对较低的HO2•,其亦会与•OH发生反应[式(4),式(5)],进而导致体系中•OH的含量降低[17].因此,本研究体系中ATZ的降解速率随H2O2浓度增大呈现出先增大后降低的变化趋势.
根据ATZ降解产物的定性分析结果,结合文献[18]及前述研究,本文提出UV/H2O2工艺中ATZ的可能反应类型.除在UV辐照工艺中的10种反应类型外,UV/H2O2工艺中的反应还包括:(11)脱羟基–脱侧链胺基反应,包括与胺基相连的侧链,生成产物如P13;(12)侧链脱氢羟基化反应,生成产物如P14和P15;(13)脱氢–烯化反应,对侧脱烷基–烷基氧化反应,生成物含有一个醛基乙酰胺,如P16.
2.2.3 UV/TiO2工艺中ATZ产物生成及机理 UV/ TiO2工艺能有效去除水中的有机污染物,包括其它工艺无法去除或去除效率较低的有机微污染物.为研究ATZ在UV/TiO2工艺中降解产物的生成规律,本文对TiO2加入量分别为5和30mg/L的ATZ水溶液进行了光照处理.结果表明,UV/TiO2工艺中,除检出P1~P9、P11外,还检出了8种新降解产物[19],将其分别标记为P17、P18、P19、P22、P23、P24、P25和P26[图4(D)],且TiO2的存在及浓度未改变ATZ降解产物的种类[图4(E)],只对产物生成量造成一定影响.TiO2的光催化机理为TiO2吸收hv,使得价带(VB)上的电子(e–)被激发形成光生电子,光生电子由VB跃迁到导带(CB),从而形成了电子–空穴对[20]. VB上e–跃迁后形成的空穴(+)可与溶液中水分子电离出的OH–结合,生成•OH和e–,•OH可将包括生物难降解有机物在内的大部分有机物彻底氧化为CO2、H2O和其他无机物.而生成的e–亦可与TiO2表面的O2结合,生成超氧化物(•O2–),由于+、•OH及•O2–的氧化性均较强,它们均可直接与ATZ发生反应.
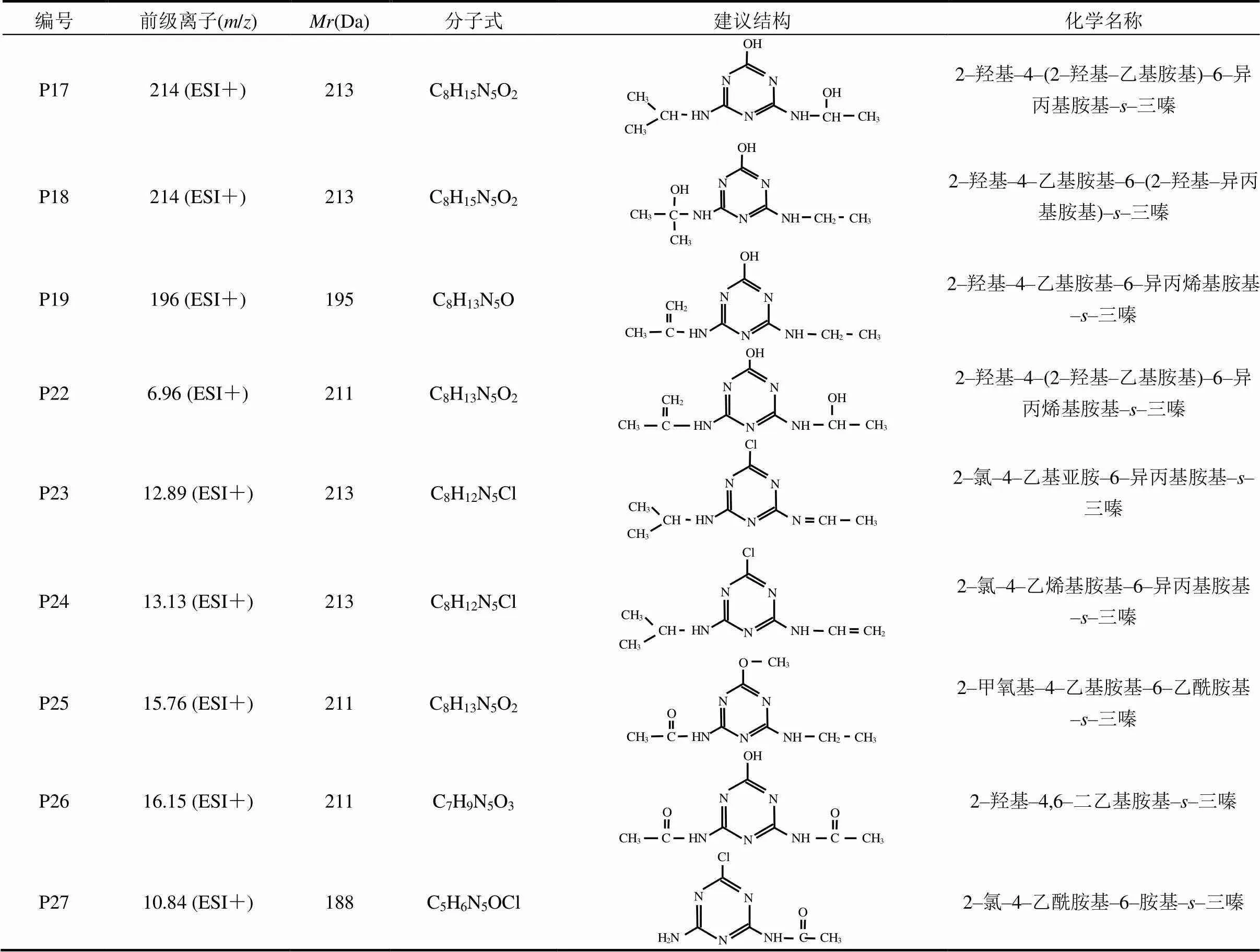
表3 UV/TiO2工艺中阿特拉津降解产物的建议分子结构式及相关信息
UV/TiO2工艺降解复杂结构有机物的过程中,往往会产生大量的中间产物.本文基于测得的质谱和色谱信息及文献[21],推导出了新检出氧化产物的分子结构(表3).根据ATZ光降解产物的定性、定量分析结果,并结合文献[22]及本研究结果,提出了UV/TiO2工艺中ATZ的降解途径.除其在单独UV辐照工艺中发生的10种反应类型外,还包括:(14)脱氢–羟基化反应,生成产物如P17、P18、P22、P24;(15)脱羟基–烯化反应,生成产物如P19、P23.
2.2.4 UV/H2O2/TiO2工艺中ATZ产物生成及机理 对H2O2/TiO2浓度分别为5mg/L和30mg/L的ATZ水溶液进行了光照处理,检测出了其在UV、UV/H2O2(5mg/L)、UV/TiO2(5mg/L)工艺中除P24外的所有产物,亦检出一新产物P27,见图4(F).研究发现, H2O2/TiO2浓度变化未改变ATZ降解产物的种类([图4(G)],只对产物生成量存在一定影响.此外,H2O2和TiO2(5mg/L)同时加入时,ATZ的降解速率较UV/TiO2(5mg/L)工艺有所增加,这是由于H2O2在光照初期经光催化后可产生ROS,故能提高ATZ的降解速率.在两种催化剂综合作用下,致ATZ的降解速率呈现出先快后慢的变化趋势.H2O2和TiO2(30mg/L)同时加入时,对于ATZ的降解起到一定促进作用,但H2O2的加入同时会造成其与ATZ竞争hν;高浓度TiO2则会显著降低溶液的透光率,导致ATZ吸收的hν数量减少.
基于本研究测得的质谱和色谱信息及文献[12,16],推导出了P27的分子结构(表3).提出了ATZ在UV/H2O2/TiO2工艺中的降解途径,除其在UV、UV/H2O2、UV/TiO2工艺中15种反应类型外,还包括:(16)单侧脱甲酰甲基,对侧脱烯烃–醛化反应,生成物含有一个甲酰甲基,生成产物如P27.
2.3 光氧化对ATZ溶液需氯量及DBPFP的影响
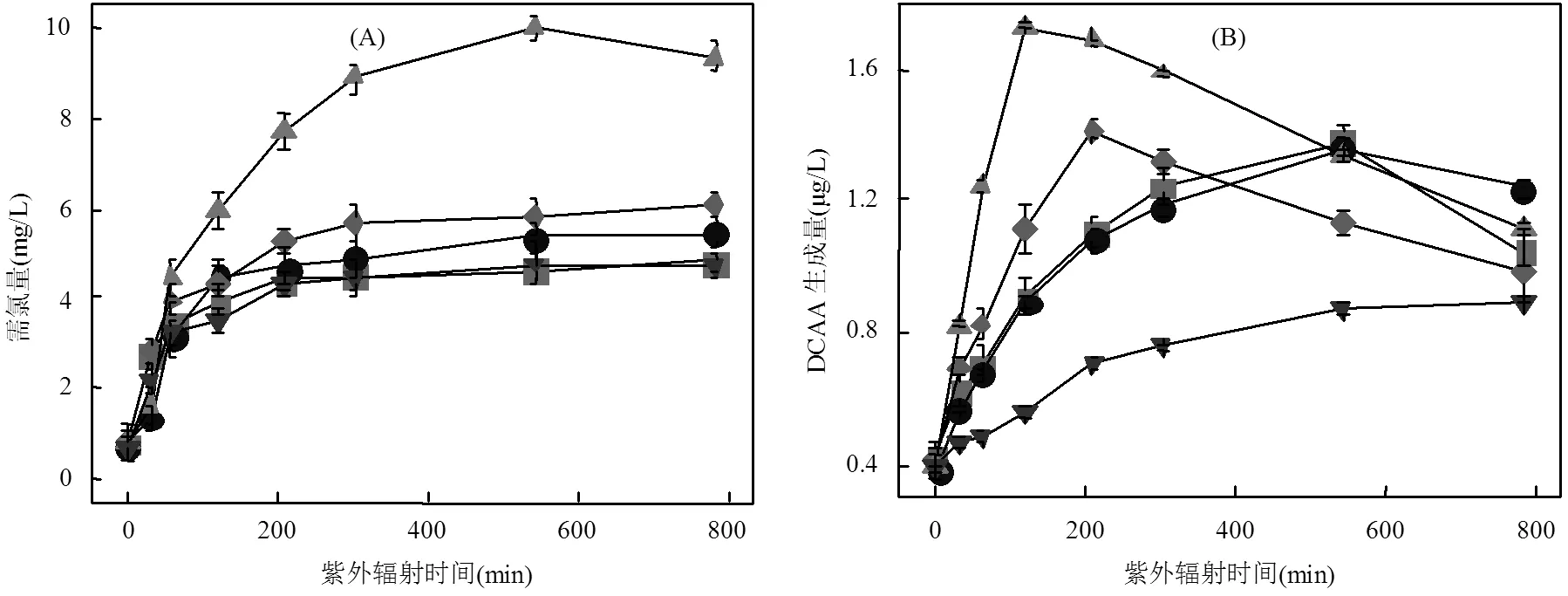
2.3.1 需氯量 需氯量与DBPs生成量存在密切关系,经不同UV辐照工艺预处理后的ATZ溶液需氯量见图5(A).由图5可知,UV光氧化预处理后,ATZ溶液的需氯量随光照时间的增加而增加,这表明ATZ部分氧化产物的生成导致溶液与氯的反应活性增强.此外,不同UV光氧化工艺处理初期,ATZ溶液的需氯量均随光照时间的增加而显著增大,一定时间后的ATZ溶液需氯量逐渐趋于稳定.这表明,ATZ在光照初期氧化生成了更易与氯反应的产物,在一定处理时间内该类产物随光照时间的增加不断积累;但光照一定时间后,易与氯反应的产物达到了动态平衡或总量较为稳定,故致ATZ溶液的需氯量变化较小.
需氯量因氧化工艺不同而不同,ATZ溶液经UV/H2O2工艺处理后的需氯量高于单独UV辐照工艺,且需氯量随体系中H2O2加入量的增加而显著增加,这是因为在氯淬灭H2O2反应中,有机物结构重组可导致新的有机物形成,这些有机物会和ATZ及其降解产物存在与氯的竞争反应,ATZ溶液在UV/ TiO2(5mg/L)工艺中的需氯量高于单独UV辐照工艺,但TiO2浓度升至30mg/L时的需氯量却出现了降低,这可能与降解产物的生成种类和浓度有关.
2.3.2 二氯乙酸 由图5(B)可知,5种条件下二氯乙酸(DCAA)的生成量均较低,且随光照时间的增加呈先增加后降低的变化趋势,DCAA生成量的降低可能与产物的进一步降解和矿化有关.ATZ溶液经UV/H2O2工艺预处理和氯化处理后的DCAA生成量高于其经单独UV辐照工艺预处理和氯化处理后的生成量,同一光照时间时,DCAA生成量随H2O2浓度增加而增加.这是由于H2O2的加入促进了体系中•OH的形成,这些ROS具有将有机物氧化降解的潜力,在此过程中生成的溶解性有机物(DOM)可能具有更高的氯反应活性,故能增加预处理后ATZ溶液的需氯量,进而增加了DBPs生成量[18].UV/TiO2(30mg/L)工艺中DCAA的生成量低于UV/TiO2(5mg/L)工艺,这可能是由于高投加量的TiO2会与UV/TiO2处理中产生的高氯反应活性DOM竞争氯,并在此过程中形成TiCl4.
2.3.3 三氯乙酸 5种条件下,三氯乙酸(TCAA)生成量均随光照时间的增加呈先增加后降低的变化趋势.ATZ经UV/H2O2工艺预处理和氯化处理后的TCAA生成量远高于经单独UV辐照工艺预处理和氯化处理后的TCAA生成量,且TCAA生成量随H2O2浓度变化而变化,这可能是H2O2浓度变化改变了与氯反应活性不同的DOM的生成量,且UV/H2O2工艺中产生的•OH能将大分子疏水性DOM转化为小分子亲水性DOM.亲水性DOM与氯的反应性一般较疏水性DOM高,故ATZ溶液经UV/H2O2工艺预处理后的DBPs前驱物增加,导致溶液的需氯量增加,这均利于氯化过程中TCAA的生成[23].如图5(C)所示,UV/H2O2(30mg/L)工艺中,TCAA生成量在120min时达到峰值,较0min增加了1043.16%. UV/TiO2工艺中TiO2的浓度对TCAA生成量的影响较小,但TiO2的存在导致体系中生成了更多的TCAA.
2.3.4 三氯甲烷 三氯甲烷(TCM)是ATZ经UV光氧化预处理及氯化处理后的主要DBPs之一,ATZ溶液经UV/H2O2、UV/TiO2工艺预处理和氯化处理后的TCM生成量,均随光照时间的增加呈现出先增加后降低的变化趋势,但达到最大生成量时所需的光照时间均低于单独UV辐照工艺. UV/H2O2工艺中,ATZ溶液的TCM生成量随H2O2浓度的增加而降低,表明低H2O2剂量能够促进体系中TCM前驱物的生成,此结果与文献[24]一致.例如,在原位氯胺法中,游离氯主要转化为氯胺,是主要的活性氯物种,TCM的降低归因于氨与游离氯的反应[23].
2.3.5 三氯硝基甲烷 5种条件下,三氯硝基甲烷(TCNM)生成量均较低,最大生成量为0~1.6μg/L.单独UV辐照工艺中TCNM生成量随光照时间的增加而呈增加趋势,UV/H2O2工艺中TCNM的生成量随光照时间的增加呈先增加后降低的变化趋势,达到最大生成量时所需要的光照时间均低于单独UV辐照工艺.这是由于UV/H2O2工艺处理可显著降解氯活性和氯胺活性的溶解有机氮(DON),而DON是亚硝胺的主要前体,故UV/H2O2工艺预处理能将后续氯化或氯胺化反应过程中的亚硝胺生成量降低50%以上.H2O2浓度由5mg/L增至30mg/L时,ATZ溶液的TCNM生成量增加39.33%,达到TCNM最大生成量时的光照时间亦缩短.经UV/TiO2(5mg/L)工艺预处理和氯化处理后,ATZ溶液的TCNM生成量随光照时间的增加呈现出先增加后趋于稳定的变化趋势;经UV/TiO2(30mg/L)工艺预处理和氯化处理后,ATZ溶液的TCNM生成量随光照时间的增加而显著增加,见图5(E).
2.3.6 三氯丙酮 三氯丙酮(TCP)是ATZ经不同UV光氧化预处理及氯化处理后的DBPs之一.5种条件下,ATZ溶液的TCP生成量均随光照处理时间的增加而增加,但其增加幅度因反应条件的不同而不同.对于单独UV辐照工艺,ATZ溶液的TCP生成量随光照时间的增加而缓慢增加; UV/H2O2工艺中,TCP生成量随H2O2浓度的增加而显著增加;UV/TiO2工艺中,TiO2浓度由5mg/L增至30mg/L时,TCP生成量增加了146.35%,这可能是由于过氧自由基之间的反应可在短时间内导致酮或醛的产生(在不完全矿化的情况下)所致,见图5(F).
4 结论
4.1 pH=7.0条件下,ATZ在4种UV光氧化工艺中的氧化降解均遵循准一级反应动力学.单独UV辐照工艺可氧化去除水溶液中的ATZ,但效果不佳;UV与H2O2(5mg/L)联用工艺可一定程度上提高ATZ的氧化降解速率;而UV/TiO2工艺则会降低ATZ的降解速率.
4.2 对于ATZ溶液,经单独UV、UV/H2O2、UV/ TiO2、UV/H2O2/TiO2工艺处理后分别检出了10、15、17、23种氧化产物;H2O2或/和TiO2浓度会影响ATZ降解速率和产物生成量,但未影响产物种类.
4.3 单独UV辐照工艺中,ATZ的去除为UV照射作用下的缓慢光解;UV/H2O2工艺中,ATZ的去除为UV光解和•OH氧化作用所致;UV/TiO2和UV/ H2O2/TiO2工艺中,ATZ的去除是UV光解和+、•OH及•O2–氧化的共同作用所致.
4.4 ATZ的不完全氧化可产生多种副产物,造成氯化DBPFP显著增大.UV光氧化初期,DBPs生成量随处理时间的增加而显著增加,TCM和TCP为主要DBPs种类.有机物的UV光氧化处理可能生成毒性较小的产物,但其在后续氯化过程中的DBPs可能显著增加.因此,采用UV光氧化工艺处理有机物时,需综合考虑其对后续工艺及出水水质的影响,并通过工艺组合及优化,在保证出水水质安全的前提下达到经济、高效的目的.
[1] Loos R, Gawlik B M, Locoro G, et al. EU–wide survey of polar organic persistent pollutants in European river waters [J]. Environmental Pollution, 2009,157(2):561–568.
[2] McClure C M, Smalling K L, Blazer V S, et al. Spatiotemporal variation in occurrence and co–occurrence of pesticides, hormones, and other organic contaminants in rivers in the Chesapeake Bay Watershed, United States [J]. Science of the Total Environment, 2020,728:138765–138777.
[3] Fang W, Peng Y, Muir D C, et al. A critical review of synthetic chemicals in surface waters of the US, the EU and China [J]. Environment International, 2019,131:104994–105004.
[4] 佘伟铎,石运刚,李 洁,等.长江流域重庆段水体和沉积物中农药分布特征及风险评价 [J]. 生态毒理学报, 2020,15(2):220–230.
She W D, Shi Y G, Li J, et al. Distribution characteristics and risk assessment of pesticides in water and sediments of Chongqing section of Yangtze River Basin [J]. Asian Journal of Ecotoxicology, 2020, 15(2):220–230.
[5] Jin Y, Wang L, Chen G, et al. Exposure of mice to atrazine and its metabolite diaminochlorotriazine elicits oxidative stress and endocrine disruption [J]. Environmental Toxicology and Pharmacology, 2014, 37:782–790.
[6] Bianchi C L, Pirola C, Ragaini V, et al. Mechanism and efficiency of atrazine degradation under combined oxidation processes [J]. Applied Catalysis B: Environmental, 2006,64(1):131–138.
[7] 胡 倩,阳 海,石 妮,等.光催化体系中噻虫胺降解动力学及机制 [J]. 环境科学, 2016,37(9):3524–3531.
Hu Q, Yang H, Shi N, et al. Degradation kinetics and mechanism of clothianidin in photocatalytic system [J]. Environmental Science, 2016,37(9):3524–3531.
[8] 刘玉灿,苏苗苗,张 岩,等.不同UV工艺中阿特拉津的降解效果与机理研究 [J]. 中国给水排水, 2019,35(5):60–66.
Liu Y C, Su M M, Zhang Y, et al. Degradation effect and mechanism of atrazine in different UV processes [J].China Water and Wastewater, 2019,35(5):60–66.
[9] Klassen N V, Marchington D, Mcgowan H C. H2O2determination by the I3–method and by KMnO4titration [J]. Analytical Chemistry, 1994,66(18):2921–2925.
[10] Liu Y, Duan J, Li W, et al. Determination of volatile disinfection byproducts in water by gas chromatography–triple quadrupole mass spectrometry [J]. Analytical Letters, 2015,48(1):188–203.
[11] 汪 力,高乃云,魏宏斌,等.饮用水中内分泌干扰物阿特拉津UV光氧化研究 [J]. 环境科学, 2006,27(6):1144–1149.
Wang L, Gao N Y, Wei H B, et al. Study on UV photooxidation of atrazine in drinking water [J]. Environmental Science, 2006,27(6): 1144–1149.
[12] Chen C, Yang S G, Guo Y P, et al. Photolytic destruction of endocrine disruptor atrazine in aqueous solution under UV irradiation: Products and pathways [J]. Journal of Hazardous Materials, 2009,172(2): 675–684.
[13] Ailton J M, Aline C B, Luis F C G, et al. The process of atrazine degradation, its mechanism, and the formation of metabolites using UV and UV/MW photolysis [J]. Journal of Photochemistry and Photobiology, A: Chemistry, 2017,347:160–167.
[14] Li J, Hu J, Xu W, et al. Hydrolysis reaction mechanism in atrazine metabolism and prediction of its metabolites’ toxicities [J]. Journal of Agricultural and Food Chemistry, 2014,62(21):4852–4863.
[15] Karin L T, Mark J B, Shane A S, et al. Transformation of atrazine, carbamazepine, diclofenac and sulfamethoxazole by low and medium pressure UV and UV/H2O2treatment [J]. Separation and Purification Technology, 2012,96:33–43.
[16] Aaron D D, Volha S K, Debbie M, et al. UV/H2O2treatment of drinking water increases post-chlorination DBP formation [J]. Water Research, 2010,44(12):3703–3713.
[17] Khan J A, He X X, Shah N S, et al. Kinetic and mechanism investigation on the photochemical degradation of atrazine with activated H2O2, S2O82–and HSO5–[J]. Chemical Engineering Journal, 2014,252:393–403.
[18] Du P, Su T, Luo X, et al. Bi–, Y–codoped TiO2for carbon dioxide photocatalytic reduction to formic acid under visible light irradiation [J]. Chinese Journal of Chemistry, 2018,36(6):538–544.
[19] Texier I, Ouazzani J, Delaire J, et al. Study of the mechanisms of the photodegradation of atrazine in the presence of two photocatalysts: TiO2and Na4W10O32[J]. Tetrahedron, 1999,55 (11):3401–3412.
[20] 李明玉,赵 倩,曾小龙,等.TiO2光电催化中光生电子降解对苯醌的行为研究 [J]. 中国环境科学, 2015,35(5):1397–1402.
Li M Y, Zhao Q, Zeng X L, et al. Study on the degradation of p- benzoquinone by photogenerated electrons in TiO2photoelectrocatalysis [J]. China Environmental Science, 2015,35(5): 1397–1402.
[21] Yang N, Liu Y P, Zhu J, et al. Study on the efficacy and mechanism of Fe–TiO2visible heterogeneous fenton catalytic degradation of atrazine [J]. Chemosphere, 2020,252:126333–126344.
[22] Hu E, Cheng H. Catalytic effect of transition metals on microwave– induced degradation of atrazine in mineral micropores [J]. Water Research, 2014,57:8–19.
[23] Ding S K, Wang F F, Chu W H, et al. Using UV/H2O2pre-oxidation combined with an optimised disinfection scenario to control Cx3r-type disinfection by-product Formation [J]. Ecology Environment and Conservation, 2019,167:115096–115118.
[24] Zhang Y M, Chu W H, Xu T, et al. Impact of pre-oxidation using H2O2and ultraviolet/H2O2on disinfection byproducts generated from chlor(am)ination of chloramphenicol [J]. Chemical Engineering Journal, 2017,317:112–118.
Ultraviolet photooxidation of atrazine and its effect on disinfection by-product formation potential.
LIU Yu-can1*, WANG Ying1, WANG Yu-xia2, ZHU Yu-liang1, JI Xian-guo1, ZHANG Yan1, DUAN Jin-ming3, LI Wei3
(1.School of Civil Engineering, Yantai University, Yantai 264005, China;2.School of Environmental and Municipal Engineering, North China University of Water Resources and Electric Power, Zhengzhou 450046, China;3.School of Environmental & Municipal Engineering, Xi’an University of Architecture and Technology, Xi’an 710055, China)., 2021,41(10):4606~4615
The degradation kinetics and mechanisms of atrazine (ATZ) in aqueous solution by various UV photooxidation processes, as well as its influence on chlorine demand and disinfection by-product formation potential (DBPFP) in the subsequent chlorination process were systematically investigated. The results showed thatthe degradation of ATZ in different photooxidation processes followed the pseudo-first-order reaction kinetics. In comparison with low removal efficiency of ATZ by UV irradiation alone, the UV/H2O2process had much higher removal efficiency, and it first increases and then decreases with increasing H2O2concentration. In UV/TiO2process, the degradation of ATZ was directly related to the production of oxidizing active species (ROS) such as holes, •OH and •O2–. In UV/H2O2/TiO2process, the degradation rate of ATZ was found higher than that in the UV/TiO2process. After UV pretreatment and the following chlorination, five disinfection by-products (DBPs) were detected in ATZ aqueous solution. Among of them, chloroform (TCM) and trichloroacetone (TCP) were the major chlorinated DBPs. The present study showed that the degradation of ATZ followed different degradation pathways in different UV photooxidation pretreatment processes, which would significantly affect the disinfection by-product formation potential (DBPPF) of ATZ solution in the following chlorination process.
atrazine;advanced oxidation process;chlorination disinfection by-products;degradation mechanism
X703
A
100-6923(2021)10-4606-10
刘玉灿(1986–),男,山东菏泽人,副教授,博士,主要从事水中有机污染物的迁移转化规律与去除方面的工作.发表论文30余篇.
2021–02–19
山东省自然科学基金项目(ZR2017BEE016);国家自然科学基金项目(51609207);烟台大学科技项目(TM17B19)
* 责任作者, 副教授, liuyucan@ytu.edu.cn
猜你喜欢
中国机械工程(2022年8期)2022-05-09
中国机械工程(2021年8期)2021-05-07
有色设备(2021年4期)2021-03-16
天然产物研究与开发(2019年10期)2019-11-05
音乐教育与创作(2019年8期)2019-05-16
中国民族医药杂志(2016年2期)2016-05-14
中国氯碱(2014年10期)2014-02-28
河南科技(2014年4期)2014-02-27
华东理工大学学报(自然科学版)(2014年2期)2014-02-27
河北大学学报(自然科学版)(2012年3期)2012-03-25
