
Whole-genome resequencing of Japanese whiting(Sillago japonica) provide insights into local adaptations
2021-10-18 00:16ZhiQiangHanXinYuGuoQunLiuShanShanLiuZhiXinZhangShiJunXiaoTianXiangGao
Zoological Research 2021年5期
Zhi-Qiang Han, Xin-Yu Guo, Qun Liu, Shan-Shan Liu, Zhi-Xin Zhang, Shi-Jun Xiao, Tian-Xiang Gao,*
1 Fishery College, Zhejiang Ocean University, Zhoushan, Zhejiang 316002, China
2 BGI-Qingdao, BGI-Shenzhen, Qingdao, Shandong 266555, China
3 Graduate School of Marine Science and Technology, Tokyo University of Marine Science and Technology, Konan, Minato, Tokyo 108-8477, Japan
4 Institute of Fisheries Science, Tibet Academy of Agricultural and Animal Husbandry Sciences, Lhasa, Tibet 850000, China
ABSTRACT The genetic adaptations of various organisms to heterogeneous environments in the northwestern Pacific remain poorly understood. Heterogeneous genomic divergence among populations may reflect environmental selection. Advancing our understanding of the mechanisms by which organisms adapt to different temperatures in response to climate change and predicting the adaptive potential and ecological consequences of anthropogenic global warming are critical. We sequenced the whole genomes of Japanese whiting(Sillago japonica) specimens collected from different latitudinal locations along the coastal waters of China and Japan to detect possible thermal adaptations.Using population genomics, a total of 5.48 million single nucleotide polymorphisms (SNPs) from five populations revealed a complete genetic break between the Chinese and Japanese groups, which was attributed to both geographic distance and local adaptation. The shared natural selection genes between two isolated populations (i.e., Zhoushan and Ise Bay/Tokyo Bay) indicated possible parallel evolution at the genetic level induced by temperature. These genes also indicated that the process of temperature selection on isolated populations is repeatable. Moreover, we observed natural candidate genes related to membrane fluidity, possibly underlying adaptation to cold environmental stress. These findings advance our understanding of the genetic mechanisms underlying the rapid adaptations of fish species. Species distribution projection models suggested that the Chinese and Japanese groups may have different responses to future climate change, with the former expanding and the latter contracting. The findings of this study enhance our understanding of genetic differentiation and adaptation to changing environments.
Keywords: Sillago japonica;Local adaptation;Climate change; Temperature stress;Wholegenome resequencing
INTRODUCTION
A fundamental question in biology is how organisms adapt to diverse and changing environments (Schluter & McPeek,2000). Spatially varying selection usually triggers differential adaptations of local populations and ultimately initiates evolutionary diversification and speciation (Ferchaud &Hansen, 2016; Fustier et al., 2017). Identification of the genetic basis for ecological adaptation is not only a primary goal of evolutionary biology but is also required to define conservation units for management (Balanyà et al., 2009;Chen et al., 2018a; Skelly et al., 2007). Moreover, detecting candidate genes under natural selection could help identify key gene pathways involved in adaptations to local environments. The marine environment is undergoing marked changes, most especially rising temperature, which is a major environmental factor that influences spatial distribution and diversity of fish species (Chen et al., 2018b). Advancing our understanding of thermal adaptation is critical in predicting the adaptive potential and ecological consequences of anthropogenic global warming.
Global climate change has considerable effects on organisms, especially coastal species. The influence of climate change on marine organisms is intensified in the East China and Yellow seas, where water is warming at a higher rate than in other areas (Cai et al., 2020). Simulation results have indicated that even under the low greenhouse emission scenario (i.e., RCP4.5), the average annual sea surface temperature in the Yellow Sea will increase by at least 2 °C by the end of the 21st century (http://www.bio-oracle.org/) (Assis et al., 2018). Therefore, climate-mediated selection signatures must be detected. Restriction site-associated DNA tag sequencing (RAD-seq) and genotyping-by-sequencing (GBS)have been applied in investigations on the genetic adaptations of organisms in the northwestern Pacific. However, these methods only cover a fraction of the total genome and may miss numerous loci under selection in local adaptations (Li et al., 2019; Wang et al., 2016; Xu et al., 2017). Furthermore,studies on population genomics conducted in this region have only revealed a possible signature of thermal adaptation, and no population-specific genomic regions or genes related to warm or cold temperature stress have been analyzed.Adaptations to high or low temperatures are expected to have highly polygenic backgrounds, which are difficult to detect using reduced representation genome sequencing. In addition to possible local adaptations, the similar distribution of ocean surface temperature between the coastal waters of China and Japan may lead to identical or similar adaptive changes in distantly independent populations, thereby causing parallel evolution.
Recent population-scale genomic study of Japanese whiting(Sillago japonica; Family Sillaginidae) suggested that it may be an ideal model for detecting signatures of parallel selection(Yang et al., 2020). This fish is a commercially important coastal species widely distributed throughout the northwestern Pacific, especially the East China Sea, Yellow Sea, and coastal waters of Japan (McKay, 1992; Oozeki et al., 1992).This species is euryhaline but does not migrate over long distances (Yang et al., 2020). GBS markers indicate that there are substantial genetic differences between the Chinese and Japanese populations, with the Rushan (RS) (Weihai City,China) population considered a transitional population between China and Japan (Yang et al., 2020), indicating that the species has dispersed from the South China Sea to the East China Sea, Yellow Sea, and coastal waters of Japan.These findings support the supposition that theS. japonicapopulations in the East China Sea and coastal waters of Japan are independent genetic populations. The Yellow Sea population, which dispersed from the East China Sea, may tolerate cold temperature stress during winter and may exhibit genomic adaptations to such temperatures. The Japanese population, which originated from the Yellow Sea, may suffer warm temperature stress. Although the East China Sea populations from western coastal China originated from the South China Sea, these populations and some Japanese populations encounter similar temperature stress, which could cause parallel evolution and natural selection on identical genes. Therefore,S. japonicais an interesting species for studies on ecological adaptation because it inhabits diverse environments, ranging from tropical to warm temperate climates, and it shows low levels of genetic differentiation at neutral loci (Gao et al., 2019; Kashiwagi et al., 2000). A draftS. japonicagenome was previously sequenced by the BGIShenzhen Company, China (unpublished data). This draft genome provides a fundamental resource for whole-genome resequencing and population genomic research.
In the present study, we sequenced the whole genomes of 49S. japonicaindividuals collected from five sites across the coastal waters of China and Japan, covering high-temperature(mean annual temperature >25 ℃), warm-temperature (mean annual temperature >19 ℃), and cold-temperature (mean annual temperature >14 ℃) areas. This study provides insights into the evolutionary history and genetic diversity ofS.japonica, as well as the mechanisms by which a species can adapt to different thermal environments, especially cold regions. By comparing genomes from low- and hightemperature environments, we identified several candidate genes with potential molecular functions in local temperature adaptations among theS. japonicapopulations inhabiting different thermal environments. Comparison ofS. japonicapopulations from the East China Sea and coastal waters of Japan may provide evidence of parallel evolution within this species.
MATERIALS AND METHODS
Sampling
We collected 49 individuals ofS. japonicafrom five populations distributed throughout the coastal waters of China and Japan (Figure 1; Table 1). All individuals were identified based on morphological features. A piece of muscle tissue was obtained from each individual and preserved in 95%ethanol or frozen for DNA extraction.
Sequencing, read mapping, and single nucleotide polymorphism (SNP) calling
Genome sequencing was performed using the Illumina HiSeq 2500 platform by Novogene Bioinformatics Technology Co.,Ltd., China. Raw read quality control and removal of potential adaptor sequences were performed to ensure the accuracy of bioinformatics analysis. Paired-end reads from each individual were aligned to the reference genome (unpublished data)using the Burrows-Wheeler Aligner (BWA) with the option“mem-t 4-k 32 -M” (Li & Durbin, 2009). After mapping, the resulting bam files were sorted using SAMtools (Li et al.,2009) and duplicate reads were removed.

Figure 1 Map of sampling locations and population genomic analyses of Sillago japonica
Table 1 Population samples of Sillago japonica used in this study
Raw SNPs were identified based on mpileup files generated by SAMtools. The filtering threshold for raw SNPs was set to a quality score of ≥20. The SNPs were discarded if their total coverage was less than one third or greater than five times the overall coverage. If two SNPs were <5 bp apart, both SNPs were removed.
Genomic distribution of genetic variants
ANNOVAR (Wang et al., 2010) was used to annotate the genomic distribution of variants and to classify them into different categories (i.e., nonsynonymous, synonymous, UTR,5 kb upstream, 5 kb downstream, intronic, and intergenic).The filters for variants were set to a minor allele frequency of >0.01, depth of >3, and missing rate of <0.2.
Population diversity and structure
We used VCFtools (Danecek et al., 2011) to estimate nucleotide diversity (window size of 40 kb) and theFSTdivergence statistic (window size of 40 kb) for each population pair. We used Plink (Purcell et al., 2007) (http://pngu.mgh.harvard.edu/~purcell/plink/) to prepare input data for Admixture (Alexander et al., 2009) (http://dalexander.github.io/admixture/index.html) to investigate population structure,with ancestry clusters ranging from two to six. Principal component analysis (PCA) was performed using GCTA software (Yang et al., 2011). The filtered SNP dataset was used to generate neighbor-joining trees with Treebest-1.9.2.Mantel tests, withFST/(1−FST) and distance matrix analysis performed using the ade4 package (Dray & Dufour, 2007) to test for isolation by distance. Geographical distances among samples were measured by following the coastline (coastal distance) and by the shortest distance across open waters(oceanic distance).
Demographic analysis
The demographic history of all five populations was analyzed using the Pairwise Sequentially Markovian Coalescent(PSMC) model in the PSMC package (Li & Durbin, 2011). In the absence of mutation rates forS. japonicaor any closely related species, we used a mutation transition matrix based on medaka data (Spivakov et al., 2014). The point mutation rate and recombination rate per base were assumed to be 2.5×10−8. Generation time was calculated asg=a+(s/(1−s)),wheresis the expected adult survival rate (assumed to be 80%) andais sexual maturation age (one year forS.japonica). A generation time of 5 years was used. To determine the variance in the estimated effective population size, we performed 100 bootstraps for each population.Population-level admixture analysis was conducted using TreeMix v.1.12 (Pickrell & Pritchard, 2012), which inferred the maximum-likelihood (ML) tree for five populations. A window size of 1 000 was used to account for linkage disequilibrium(-k) and “-global” to generate the ML tree. Migration events(-m) were sequentially added to the tree.
Screening for selection signatures
To uncover the genetic variants involved in local adaptation of each population, we calculated the genome-wide distribution ofFSTvalues and π ratios for four control-thermal groups.These groups included the cold-temperature population (RS)vs. control groups (ZS and ST), China warm-temperature population (ZS) vs. control group (RS), and Japan warmtemperature populations (IB and TB) vs. control group (RS).FSTand π for sliding windows were calculated using VCFtools(Danecek et al., 2011), with a window size of 40 kb and a step size of 20 kb. The windows with the top 5% of values for theFSTand π ratios simultaneously served as the candidate outliers under strong selective sweeps. All outlier windows were assigned to their corresponding SNPs and genes.Overlapping genes in cold and warm groups were selected for further analysis to avoid false positives. Selected genes enriched in Gene Ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways were determined using OmicShare tools (http://www.omicshare.com/tools). Multiple comparisons were corrected using the false discovery rate (FDR-adjustedP-value <0.05).Additionally, PCA was performed to infer the population structure with neutral SNP loci after removing outlier loci.
Distribution prediction of Chinese and Japanese populations under climate change
We used the maximum entropy (Maxent) model (Phillips et al.,2006) to predict potential distributions of two independent populations (Chinese and Japanese) under changing climates.We retrieved species presence records from Zhang et al.(2019) and divided them into Chinese (22 records) and Japanese (23 records) groups. Based on the results of Zhang et al. (2019) and the benthic life type ofS. japonica, we considered five environmental predictors from Bio-ORACLE(http://www.bio-oracle.org), including ocean depth, distance to shore, mean sea benthic temperature, salinity, and current velocity. The five predictors were not highly correlated (i.e.,pairwise Pearson correlation coefficients |r|<0.70) and thus all were used to develop the Maxent models. Future (i.e., RCP4.5 in 2100s (average from 2 090 to 2 100)) marine environmental predictors, including temperature, salinity, and current velocity,were also downloaded from Bio-ORACLE. We assumed that ocean depth and distance to shore would remain unchanged in the future. For each group, we developed a Maxent model using all presence data of the group and present-day environmental predictors; the model was further used to predict potential distributions under present-day and future climatic conditions.
RESULTS
Sequence information and genetic structure
We sampled 49 individuals from five populations covering the East China Sea, Yellow Sea, and Pacific coastal waters of Japan (Figure 1A; Table 1). Whole-genome resequencing yielded 680 Gb of sequencing data and generated 13.6 Gb for each individual. The whole-genome sequence raw reads forS.japonicawere deposited in the NCBI Sequence Read Archive under accession No. PRJNA743415. Genome alignment resulted in an average depth of 20.91-fold and average genome coverage of 99.40% (at least one-fold)(Supplementary Table S1). All individual data were aligned to the reference genome (mapping rate from 95.00% to 96.89%)(Supplementary Table S2), and SNPs were called after rigorous quality filtering. We identified 5 483 086 SNPs after quality control for further analysis, including 406 925 in exonic regions, 2 025 230 in intronic regions, and 2 649 227 in intergenic regions. Of the exonic SNPs, we identified 312 482 synonymous and 94 443 nonsynonymous SNPs(Supplementary Table S3). The nucleotide diversity π for each population was similar, ranging from 1.64×10-3to 1.69×10-3(Figure 1B).
Clustering analysis was performed using PLINK and Admixture to examine the relationships among populations(Figure 1C). Admixture analysis revealed clear evidence of clustering. AtK=2, the admixture structure showed two clusters, with populations from China (RS, ZS, and ST)forming one cluster and populations from Japan (IB and TB)forming another. AtK=3, the RS population formed a separate cluster. As theKvalue increased, the ST population separated as a unique cluster. WhenKincreased from 3 to 6, the ZS population was shared with clusters from RS and ST. Both TB and IB were highly admixed in all cases withKfrom 2 to 6.The PCA results recovered similar clusters as the admixture analyses (Figure 1D). The first principal component (PC1)separated the Chinese and Japanese clades, consistent with the admixture result atK=2. The second principal component(PC2) further separated the RS, ZS, and ST populations but was not able to distinguish between the TB and IB populations, consistent with the admixture results atK=5 and 6 (Figure 1C). All individuals from the Chinese groups clustered within the population defined by their sampling location, revealing an obvious geographic structure signal from the East China Sea to the Yellow Sea.
The phylogenetic tree further supported the admixture and PCA results. A neighbor-joining tree was constructed based on whole-genome SNPs (Figure 1E). The tree formed two clades that represented geographic divergence between the coastal waters of China and Japan. In the Chinese clade,three distinct clusters generally defined by geographic localities were recovered. However, the phylogenetic topology of the Japanese clade showed a shallow structure, and some individuals were grouped into another geographic population.
We calculated pairwiseFSTbetween populations to quantify their genetic differentiation (Table 2). PairwiseFSTranged from −0.000 1 to 0.037 7, with an average of 0.022 4,consistent with the overall population structure. SignificantFSTvalues between the Chinese (RS, ZS, and ST) and Japanese populations (IB and TB) ranged from 0.023 7 to 0.037 7, with an average of 0.0316. The level of genetic differentiation among Chinese populations was higher than that between the two Japanese populations.
Isolation by distance
To explore the influence of geographic distance on genetic differentiation, we performed Mantel tests to determine the association between the geographic distance matrix and pairwiseFSTmatrix. Two geographic distance patterns (i.e.,coastal and oceanic distances) were used in the tests(Figure 2A, B). We identified a closer relationship (r=0.90,P=0.000 2) betweenFST/(1−FST) and coastal distance than that betweenFST/(1−FST) and oceanic distance (r=0.80,P=0.002 9). This indicated isolation due to coastal distance, with coastal distance explaining 91% of the variation in genetic differentiation for the species. Thus, population structure analyses and Mantel tests collectively indicated that strong barriers may have had a greater influence on population differentiation than other factors.

Table 2 Pairwise FST values for five populations
Demographic history and migration between populations
The demographic history ofS. japonicawas inferred to understand its evolutionary history. Historical effective population sizes were estimated via PSMC. Results showed that the demographic history ofS. japonicacould be traced to~100 thousand years ago (Ka), and the Chinese and Japanese populations experienced markedly different demographic histories (Figure 2C). The three Chinese populations experienced similar demographic trajectories, with one large population expansion and one population bottleneck. The Chinese populations showed an obvious population expansion ~30 Ka. The effective population size of the Chinese populations peaked ~2-10 Ka, and then declined to a bottleneck ~2 Ka, especially the RS population. Among the three Chinese populations, the population expansion signals increased from north to south, but the bottleneck signal decreased from north to south. The Japanese IB and TB populations showed a different demographic history than the Chinese populations, with only one small population expansion. In contrast to the large and rapid population expansion of the Chinese populations, the Japanese populations started to increase slowly ~10 Ka and reached a peak in present-day.
We reconstructed the ML tree of the five populations using TreeMix to address the population history relationships and identify pairs of populations related to each other independent of that captured by the tree (Figure 2D). The ML tree without migration events inferred from TreeMix analysis divided the 49 individuals into two clusters, similar to the population structuring patterns identified from phylogenetic tree analysis,PCA, and genetic structure analysis. When potential migration edges (i.e., migration events between branches) were added to the ML tree, strong migration events were detected between the clusters (Figure 2E). We observed a significant migration edge (P<2.2E-308) with an estimated weight of 28.8%, which provided evidence of gene flow from the Japanese TB population to the RS population.
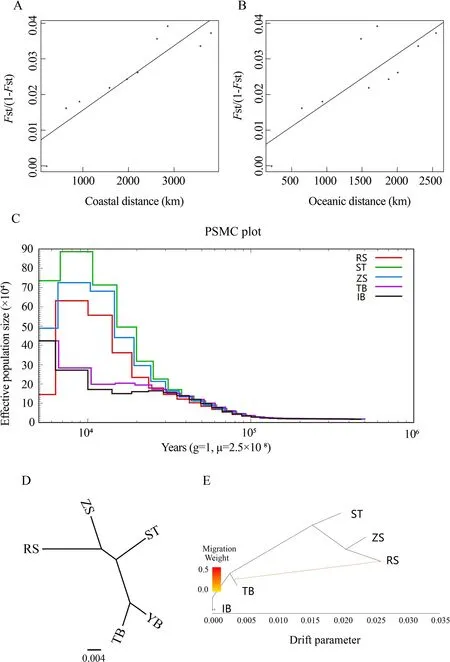
Figure 2 Isolation by distance, demographic history, and pattern of population splits
Genome-wide selection pressure analysis
We compared the genomes ofS. japonicaindividuals to identify signatures of positive selection under different environmental selection pressures. Considering that genetic isolation occurred between the Chinese and Japanese populations, we chose the ST and ZS populations as control groups to reveal candidate cold-temperature selection genes in the RS population. Using the top 5% of maximumFST(FST≥0.126 0) and π ratio (ΠST/RS≥1.238 0) values, a total of 132 candidate genes (corresponding to 3.64 Mb in size) were identified in the RS population when the ST population was used as the control group (Figure 3A). When the ZS population was used as the control group, 509 candidate genes were identified (corresponding to 18.96 Mb) (FST≥0.090 4, ΠZS/RS≥1.267 6). To validate the candidate genes under strong selective sweeps in the RS population, we identified a total of 81 overlapping genes in the RS/ZS and RS/ST pairs as potential genes associated with coldtemperature adaptation for subsequent analyses (Figure 3B;Supplementary Table S4).
To detect possible parallel adaptation between the ZS and IB/TB populations, we identified 692 genes in ZS and 122 genes in the Japanese groups involved in warm-temperature adaptation, with the cold-temperature RS population used as the control group (Figure 3C). Furthermore, 111 of the 122(91.0%) warm-temperature adaptation genes in the Japanese populations overlapped with the warm-temperature adaptation genes in the ZS population (Supplementary Table S5).Considering the geographic isolation based on whole-genome SNPs and similar temperature environments, the highly shared selection genes between the Japanese and ZS populations suggest possible parallel adaptive evolution.

Figure 3 Genomic regions with strong selective signals in populations of S. japonica
The PCA results based on SNPs of candidate genes related to cold-temperature adaptation indicated that individuals from the RS population were separated from other populations(Figure 4A). PC1 tended to separate populations with different latitudes from south to north, while PC2 separated RS from the other populations. The allele frequency of one SNP within the cold-temperature adaptation genePicalmwas significantly associated with population temperature (Figure 3D). However,the allele frequencies of one SNP within the warmtemperature adaptation geneSORCS3showed high values in the three warm-temperature populations (Figure 3E).
The PCA results based on SNPs of candidate genes related to warm-temperature adaptation also divided the 49 individuals into two clades between the ST population and the other four populations, with the RS population at the intermediate position (Figure 4B). This topological pattern was different from that inferred from the genome-wide SNPs(Figure 1D), thus supporting the credibility of the candidate genes under selection. The PCA substructure based on the candidate parallel genes related to warm-temperature adaptation revealed a closer relationship between the ZS populations and IB/TB pair than with genome-wide SNPs(Figure 4B). We filtered the SNP data after removing outlier loci from both cold and warm adaptive regions. The PCA results based on neutral SNP loci were similar to that obtained from the total loci (Supplementary Figure S1).
To obtain a broad overview of the molecular functions of these genes and detect the most differentiated regions of theS. japonicagenome, we performed GO and KEGG enrichment analyses on the candidate selection genes. These analyses offered clear insight into the genetic evolution and adaptive mechanisms ofS. japonicaunder different thermal environments.
Functional enrichment indicated that the candidate genes under cold selection were significantly enriched in only one KEGG pathway, i.e., choline metabolism in cancer (ko05231,FDR-adjustedP=0.042 1) (Figure 4C; Supplementary Table S6). However, GO term enrichment analysis of the 81 candidate genes under cold-temperature selection classified the genes into 47 categories, including metabolic processes,biological regulation, response to stimulus, and signaling process in the biological process, membrane and membrane part in the cellular component, and binding and catalytic activity in the molecular function (Supplementary Figure S2).We found 50 significantly enriched GO terms after FDR correction (Supplementary Figure S3 and Table S7). The significantly enriched GO categories were primarily associated with cell part, cation transmembrane transporter activity, tissue homeostasis, transport, and lipid metabolism.
In total, 111 genes were identified for parallel adaptations in the ZS, IB, and TB populations. These genes were classified into 48 categories (Supplementary Figure S4), including 19 significantly overrepresented GO categories detected for thermal adaptation (Supplementary Table S8). The GO clusters were primarily enriched in cell projection, structure of the cytoskeleton, binding, and maintenance of the membrane(Supplementary Figure S2). Although no significantly enriched KEGG pathways were detected, the top 20 enriched pathways were related to metabolism (synthesis and degradation of ketone bodies, cyanoamino acid metabolism, linoleic acid metabolism, alpha-linolenic acid metabolism, and butanoate metabolism), circulatory system (vascular smooth muscle contraction), and endocrine system (salivary secretion and pancreatic secretion) (Figure 4D; Supplementary Table S9).Enrichment of the selected genes in the warm-temperature population suggested a parallel adaptive mechanism to cold temperature.

Figure 4 PCA based on SNPs located in candidate genes and top 20 enriched KEGG pathways in candidate genes
Distribution prediction of Chinese and Japanese populations by Maxent model
Based on receiver operating characteristic (ROC) analysis, the area under the ROC curve (AUC) results (0.983 in Chinese group, 0.995 in Japanese group) suggested that the Maxent model showed good predictive performance. The Maxent predictions for the twoS. japonicagroups suggested that the potential distribution of this species may change substantially(Figure 5). The predictions showed that large areas of coastal China, central Japan, and Korea were suitable for the Chinese group (Figure 5A). However, more suitable distribution areas for the Japanese group were only in southern and central Japan (Figure 5E). The northern boundary for the potential population distribution of the Japanese group in the East China Sea was Zhoushan. Climate change is predicted to influence the two groups differently. Under a future RCP4.5 scenario, habitat suitability for the Chinese group would clearly increase from south to north and potential distribution may shift northward along the coastal waters of China and Japan by 2 100 (Figure 5B, G). However, results also revealed a reduction in the potential area for the Japanese group and no clear increase in areas of northern Japan by 2 100 (Figure 5F,G). This predicted reduction in habitat suitability may indicate lower evolutionary potential to adapt to changing environments. The response curves to temperature suggested different temperature adaptations between the two groups,with the Chinese group preferring high temperatures and the Japanese group favoring low temperatures (Figures 5D, H).
DISCUSSION
New insight on genetic structure of S. japonica
Both historical and current variable environments have had profound effects on the genetic variation of species. Analyses at the genomic level can provide detailed information on the genetic structure, population history, and adaptation of species to various environments, which can facilitate their protection and management (Li et al., 2019). Here, based on population genomics, we delineated the genetic characteristics ofS. japonicapopulations via whole-genome sequencing. To the best of our knowledge, this is the first study to adopt whole-genome sequencing to assess population differentiation and selection signatures in a marine fish species in the northwestern Pacific. Population history analyses suggested that the historical sea level had a substantial influence on the effective population size of this species, with a warmer climate facilitating population growth.We also identified several genes related to local environment adaptation.

Figure 5 Predicted potential distribution (A, B), changes in habitat suitability (C) of Chinese group under RCP45 scenarios, and response curves of predicted occurrence probability (D) of Chinese group against temperature. Predicted potential distribution (E, F), changes in habitat suitability (G) of Japanese group under RCP45 scenarios, and response curves of predicted occurrence probability (H) of Japanese group against temperature
Our study also provided a higher population structure resolution than that found in previous studies based on GBS,mtDNA control region, and morphological data (Gao et al.,2019; Xue et al., 2010). For example, using morphological analysis ofS. japonica, Xue et al. (2010) found no significant differentiation among populations from the Yellow Sea, East China Sea, and South China Sea. In addition, using the mtDNA control region to explore genetic differentiation, Gao et al. (2019) detected no genetic structure inS. japonicadue to the short fragment of this region. Using GBS technology, Yang et al. (2020) observed considerable genetic differentiation between the Chinese and Japanese populations ofS. japonicabut failed to separate individuals from these populations. In contrast to the mtDNA and GBS results, our study revealed a complete genetic break between the Chinese and Japanese populations according to whole-genome sequencing data.Furthermore, PCA distinguished the three Chinese populations.
Pleistocene glaciations are considered important events that shaped the phylogeographic genetic structures of extant species (Han et al., 2018). The divergence between the Chinese and Japanese populations may reflect historical isolation between the East China Sea and Pacific Ocean during the Pleistocene low sea-level stands. In the present study, population demographic analysis suggested a divergence time of 30 Ka between the two clades during the last glacial period. The date of divergence was consistent with geological events that may have created a vicariant barrier between theS. japonicapopulations of the Pacific Ocean and East China Sea (Kawahata & Ohshima, 2004).
Mantel tests identified a strong relationship between coastline distance and genetic differentiation. The ocean distance that separates the western and eastern East China Sea was identified as a physical barrier that restricted gene flow between samples. Considering the life history of this species and the physical environment in the East China Sea,long ocean distance is a reasonable physical barrier for a demersal species. Furthermore, ocean depth (<30 m) is known to limit the distribution ofS. japonicain marine waters(https://www.fishbase.in/summary/Sillago-japonica.html). The average depth of the East China Sea is about 370 m, with a maximum of 2 719 m at the continental slope (Guan & Mao,1982). Therefore, the depth of water along the direct dispersal route between the coastal waters of China and Japan may have formed an unsuitable habitat for this species and prevented its offshore dispersal. Historical migration events from the TB to RS populations and lack of migration from the Japanese populations to the ZS population support a coastal dispersal pattern. This pattern is also supported by the potential distribution areas of the two groups predicted by the Maxent model. A coastal dispersal pattern has also been observed in Japanese grenadier anchovy (Coilia nasus) (Gao et al., 2014; Han et al., 2015), a species with similar biological characteristics and geographical distribution in the East China Sea asS. japonica. Notably, amplified fragment length polymorphism (AFLP) and mtDNA analysis ofC. nasussuggested that direct ocean distance with deep water at the continental slope between the western and eastern coastal waters of the East China Sea served as a major physical barrier to this species.
The different demographic histories between the Chinese and Japanese populations may have resulted from geographic and climatic differences. The three Chinese populations showed an obvious and rapid population expansion ~30 Ka. In contrast, the Japanese populations started to increase slowly~10 Ka, reaching a peak at present-day. Moreover, the TB and IB populations showed a slower rate of increase compared with the Chinese populations over the same time.The coastal waters of Japan, with no serious historical sealevel rises, experienced less impact during the Pleistocene glaciations. The expansions of the Chinese and Japanese populations are consistent with historical sea-level increases during the interglacial periods. Notably, the populations started to increase dramatically ~30 Ka when the Wurm glacial stage began to end in the northwestern Pacific (Kawahata &Ohshima, 2004).
Pelagic and demersal fish in the coastal waters of China are expected to exhibit little intraspecific genetic structuring due to the ocean currents and apparent lack of physical barriers (Han et al., 2008). Given its wide distribution and ecological characteristics,S. japonicamay be susceptible to heterogeneous local environments. Likewise, genome-wide SNP studies have revealed remarkable regional differentiation between northern and southern populations of rough-skin sculpin (Trachidermus fasciatus), marbled rockfish(Sebastiscus marmoratus), spotted seabass (Lateolabrax maculatus), and small yellow croaker (Larimichthys polyactis)(Li et al., 2019; Liu et al., 2016; Xu et al., 2017). Thus, due to local adaptations to heterogeneous environments, a high degree of regional differentiation between northern and southern Chinese populations may be common in marine fish.For instance, average annual sea surface temperatures range from 15.9 °C in the RS population to 25.2 °C in the ST population (data provided by the Bio-ORACLE). This obvious difference in the thermal environment may have resulted in divergent selection on specific genes, and genetic diversity in certain genomic regions would be substantially decreased due to natural selection.
Adaptive mechanisms for cold temperature
Understanding how ecological variants of fish can affect population structure will have comprehensive implications for conservation management and decision-making (Li et al.,2019). Water temperature is one of the most important abiotic factors that influence the phenotypes and habitats of aquatic organisms (Chen et al., 2018b). As an important environmental stressor, low temperatures can have broad biological effects on marine organisms. These thermal effects can generate intense selective pressure on genes and genomic regions.
In the current study, selective sweep analysis was conducted to identify thermally affected genes between the northern and southern populations ofS. japonica. Combining alternative statistical approaches to detect selection signatures can provide more robust results by decreasing false-positive rates. We evaluated the genetic attributes of candidate genes relative to the genomic background. We scanned the genome-wide variations usingFSTvalues and π ratios.
A total of 81 candidate genes in the RS population identified by both π ratio andFSTanalyses were recognized as potentially affected genes related to cold adaptation. Cold adaptation and acclimatization studies suggest that more than one mechanism is involved in the biological response to cold stress (Liu et al., 2018; White et al., 2012). As expected,based on the biological complexity of cold adaptation, several different processes, rather than one term or pathway, were identified by our selection tests. Low temperatures can influence energy metabolism. Here, four selection genes (lowdensity lipoprotein receptor (LDLR),ZBTB20,PICALM, andnup35) were related to the lipid metabolic process. Lipids are the main components of the cytomembrane (van Meer et al.,2008). A common cold adaptation mechanism for the cell is to manipulate the membrane lipid composition to maintain membrane fluidity and, correspondingly, proper membrane permeability and function of protein complexes (e.g.,transporters) (Russell & Nichols, 1999).
Previous studies have shown that cell membrane permeability, integrity, and stability can be damaged under long-term low-temperature conditions (Cardona et al., 2014;Liu et al., 2018).LDLRis an important candidate gene for cold adaptation. As a glycoprotein located on the surface of cells,LDLRmediates the endocytic uptake of low-density lipoprotein(LDL) cholesterol in the liver and is a key metabolic regulator of plasma LDL cholesterol (Yamamoto et al., 1984).LDLRis the most powerful determinant of variation in total and LDL cholesterol levels (Hansen et al., 1997). This gene also plays an essential role in protecting cell membrane integrity under cold stress and strong selection pressure on this gene is useful for low-temperature adaptation (Dong et al., 2013).Similar results have also been observed for the low-density lipoprotein receptor-related protein 5 (LRP5) gene (Cardona et al., 2014).LRP5shows strong signals of selection in indigenous Siberian human populations, exhibits high expression in the liver, and plays a role in cholesterol metabolism. Natural selection of theLRP5gene helps Siberians to cope with cold climates (Cardona et al., 2014). An elevated basal metabolic rate is also a cold adaptation strategy. The selection geneTPOis involved in thyroid metabolism (Leonard et al., 2005), which determines the basal metabolic rate of the body. Thus, natural selection of theTPOgene in the RS population may maintain the basal metabolic rate under cold stress.
We also detected substantial adaptive evidence concerning ion exchange and transportation, which are processes that affect the fluidity and permeability of the cell membrane and are directly and indirectly linked to thermal regulation(Maksimov et al., 2017). We identified numerous genes that encode transporters (e.g., MFS transportersSLC22A5,SLC7A2, andSLC25A5) and ion channels (e.g., voltage-gated sodium channelSCN4B) in genomic regions of the RS population under selective sweeps. For example, we identified four copies ofSLC22A5in the genome ofS. japonica, thus indicating gene expansion.SLC22A5is a specific transporter found in cell membranes and mitochondria and is involved in the uptake and release of carnitine (Wu et al., 1999). Carnitine is a carrier of long-fatty acids and facilitates their transport into the mitochondria for lipid oxidation. DefectiveSLC22A5causes systemic carnitine deficiency, resulting in metabolic decompensation (Hu et al., 2018). Furthermore, theSLC7A2gene functions as a permease involved in transporting cationic amino acids across the plasma membrane (Closs et al.,1997). These transporter and ion channel genes are crucial for transmembrane transport to maintain stability between the internal and external environments of cells under cold temperature. Therefore, genes that encode transcellular ion transporters and channel proteins can be reshaped by natural selection under cold stress environments.
Smooth muscle contraction, which includes vasoconstriction and vasodilation, is also implicated in cold adaptation(Cardona et al., 2014). We identified two genes involved in this process that showed evidence of strong selection signals,i.e.,CPI17(protein phosphatase 1 regulatory subunit 14A)andCACNB4.CPI17is an inhibitor of PPP1CA and shows >1 000-fold inhibitory activity when phosphorylated, creating a molecular switch for regulating the phosphorylation of PPP1CA substrates and smooth muscle contraction in the absence of increased intracellular Ca2+(Li et al., 2001). TheCACNB4gene encodes a calcium channel subunit expressed in the heart and increases the peak calcium current, which is important for cardiac muscle contraction under cold stress(Rouhiainen et al., 2016). Although no heart rate data are available for the studied populations, cold exposure is known to increase cardiac pressure. Thus, efficient cardiovascular regulation may be a possible cold adaptive mechanism in the RS population. These genetic changes may facilitate the adaptation and survival ofS. japonicain low-temperature areas.
Cell repair and clear necrotic organelles are also important processes of cold adaptation, as found in the RS population.We identified two important genes under natural selection involved in cell repair. Interferon regulatory factor 1 (IRF1) is a transcriptional regulator that displays remarkable functional diversity in regulating cellular responses (Oshima et al., 2004).Under cold stress,IRF1causes cells to suspend proliferation to survive adverse environmental conditions and triggers apoptosis when cell damage becomes irreparable, thereby preventing harmful cells from damaging normal cells. DnaJ homolog subfamily C member 10 is involved in the correct folding of proteins and the degradation of misfolded proteins(Oka et al., 2013). It also promotes the apoptotic signaling pathway in response to endoplasmic reticulum stress.
Functional enrichment analysis identified 50 significant GO terms and one KEGG pathway after FDR adjustment. The enriched categories and pathways were primarily associated with cell part, transmembrane transporter activity, tissue homeostasis, lipid metabolism, apoptosis, and vascular smooth muscle contraction. These functional clusters are biologically relevant to cold adaptations and essential in regulating mechanisms for fish to respond to cold environments.
Evidence of parallel evolution in several warmtemperature adaptation genes
In general, geographically distinct populations exposed to similar environmental conditions evolve similar genotypic and phenotypic traits. The replicated nature ofS. japonicaprovides clear molecular signatures that can be used to recover alleles consistently associated with parallel warm-temperature adaptations between the ZS and IB/TB populations, despite different historical population origins. In the present study,most local adaptation genes (91.0%) in the Japanese populations were shared with selection genes in the ZS population, with the populations spanning long geographic distances but showing similar annual temperatures (19.1-21.3 ℃). The integration of shared selection genes and similar temperature environments could be considered as possible evidence for parallel evolution inS. japonica. The PCA results based on the SNPs of the shared selection genes revealed close but distinguishable patterns between the ZS and two Japanese populations, indicating independent selection events on the same genes between the ZS and IB/TB populations. Similarly, fixation of different loci at the same gene has been reported in marine-freshwater divergence of the three-spine stickleback (Barrett et al., 2008). Evidence of parallel evolution atTSHR, a major gene associated with reproductive timing, has also been observed between Atlantic herring populations from both sides of the Atlantic Ocean(Lamichhaney et al., 2017). Parallel genetic evolution has also been reported in the Atlantic cod andSebastiscus marmoratus(Bradbury et al., 2010; Xu et al., 2019).
Here,S. japonicawas studied to explore the genetic basis of repeated parallel evolution and to identify local adaptation genes in geographically distant and independent populations.Candidate genes were identified within peaks of parallel divergence between the ZS and IB/TB populations. These candidate genes may be important for local adaptation.Furthermore, GO clusters were primarily enriched in categories related to cell projection, structure of the cytoskeleton, protein binding, maintenance of membrane, and cell differentiation. In addition, although no KEGG pathway showed significant enrichment, the top 20 enriched KEGG pathways were related to metabolism (synthesis and degradation of ketone bodies, cyanoamino acid metabolism,linoleic acid metabolism, alpha-linolenic acid metabolism, and butanoate metabolism), circulatory system (vascular smooth muscle contraction), and endocrine system (salivary secretion and pancreatic secretion).
As subtropical/tropical originS. japonicais adapted to higher temperatures, warm temperature exerts relaxed selection pressure on genes involved in heat stress responses. Moreover, warm temperatures would likely promoteS. japonicagrowth by increasing cell membrane fluidity, rapid cell division, and extracellular/intracellular substance exchange. In the current study, among the candidate genes for warm-temperature adaptation, four genes were related to cell division (GO: 0051301), 17 genes were related to cytoskeleton (GO: 0016328), 10 genes were related to transmembrane signaling receptor activity (GO: 0004888),and eight genes were related to ion transmembrane transport(GO: 0034220). Various receptors in the membrane and ion transmembrane transport genes play important roles in the material exchange between the internal and external environments of cells to satisfy rapid cell division. We also identified three genes (FMN2,MGMT, andPOLH) that were related to DNA repair (GO: 0006281), thus helping to avoid DNA replication errors.FMN2plays a role in responding to DNA damage, cellular stress, and hypoxia by protecting CDKN1A against degradation.FMN2also promotes the assembly of nuclear actin filaments in response to DNA damage to facilitate the movement of chromatin and repair factors after DNA damage (Yamada et al., 2013).MGMTis crucial for genome stability. It repairs the naturally occurring mutagenic DNA lesion O6-methylguanine back to guanine and prevents mismatch and errors during DNA replication and transcription (Kawate et al., 2000).POLHserves as a DNA polymerase specifically involved in DNA repair by translesion synthesis (Masutani et al., 1999). Warm temperature promotes material exchange, DNA replication, and cell division. Furthermore, suitable temperature ensures that enough food resources are available in the environment.Hence, the above terms are functionally necessary for the adaptations ofS. japonicato warm temperatures.
Different climate resilience between Chinese and Japanese groups
Natural selection analysis demonstrated that the different populations exhibited different temperature adaptability at the genetic level. We also obtained evidence of differences in temperature adaptation between the Chinese and Japanese populations at the macro level through species distribution models. The differences in the potential distributions of the two groups under future climate change may reflect the different thermal abilities between the groups. The Chinese group contained different thermally adapted populations, which may possess strong adaptive capacities to future climate change.However, the Japanese group with stenothermal characteristics showed weak adaptive capacity to climate change. Our findings are different from the predicted distribution of this species reported by Zhang et al. (2019),who did not consider genetic differences within this species.Cryptic diversity clearly affects global climate change projections. Bálint et al. (2011) suggested that without discerning intraspecific genetic variation and cryptic diversity,the effects of global climate change are likely to be drastically underestimated. From our findings, the phylogeographic data and species distribution models provide a new understanding of the evolutionary trajectories of species under changing environmental conditions.
CONCLUSIONS
Based on the neutral and total SNPs, we identified two isolated populations that showed strong parallel evolution.This genetic parallelism was likely due to the similar local temperature environments, notwithstanding the different thermal origins of the populations. The population-specific genes related to adaptations to low temperatures may be considered as potential candidates for further exploration of the underlying mechanisms involved in confronting environmental challenges. In conclusion, by comparing the whole genomes ofS. japonicapopulations from different temperature regions, we revealed various genes associated with local adaptations to cold temperature environments.Specifically, candidate genes were functionally related to membrane fluidity in cold environments. These results advance our understanding of the underlying genetic mechanisms that allow fish, especially small ruminants, to adapt to extreme environments.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
S.J.X. and T.X.G. conceived and supervised the study. T.X.G.performed sample collection. X.Y.G. conducted DNA extraction and genome sequencing. Q.L. conducted SNP calling and genetic diversity analysis. S.S.L. performed PCA.Z.Q.H. analyzed the population genetic structure,demographic history, and selection signatures, and wrote the draft manuscript. Z.X.Z performed species distribution model analysis. All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Sheng-You Xu (Zhejiang Ocean University) and Fang-Rui Lou (Ocean University of China) for valuable suggestions and comments.

- Zoological Research的其它文章
- Chromosome-scale genome assembly of brownspotted flathead Platycephalus sp.1 provides insights into demersal adaptation in flathead fish
- Captopril alleviates lung inflammation in SARS-CoV-2-infected hypertensive mice
- Role of juvenile hormone receptor Methoprene-tolerant 1 in silkworm larval brain development and domestication
- Deletion of phosphatidylserine flippase β-subunit Tmem30a in satellite cells leads to delayed skeletal muscle regeneration
- Global view on virus infection in non-human primates and implications for public health and wildlifeconservation
- Prosecution records reveal pangolin trading networks in China, 2014-2019