
Comparative mitogenome phylogeography of two anteater genera (Tamandua and Myrmecophaga;Myrmecophagidae, Xenarthra): Evidence of discrepant evolutionary traits
2021-10-18 00:16ManuelRuizGarcDanielPinillaBeltrOscarMurilloGarcChristianMiguelPintoJorgeBritoJosephMarkShostell
Zoological Research 2021年5期
Manuel Ruiz-García, Daniel Pinilla-Beltrán, Oscar E. Murillo-García, Christian Miguel Pinto, Jorge Brito,Joseph Mark Shostell
1Laboratorio de Genética de Poblaciones Molecular-Biología Evolutiva, Departamento de Biología, Facultad de Ciencias, Pontificia Universidad Javeriana, Bogotá DC 110231, Colombia
2 Grupo de Investigación en Ecología Animal, Departamento de Biología, Facultad de Ciencias Naturales y Exactas, Universidad del Valle,Apartado Aéreo, Cali 25360, Colombia
3 Instituto de Ciencias Biológicas, Escuela Politécnica Nacional, Quito 17012759, Ecuador
4 Instituto Nacional de Biodiversidad (INABIO), Quito 170135, Ecuador
5 Math, Science and Technology Department, University of Minnesota Crookston, Crookston, MN 56716, USA
ABSTRACT The species within Xenarthra (sloths, anteaters, and armadillos) are quintessential South American mammals. Of the three groups, Vermilingua(anteaters) contains the fewest extant and paleontological species. Here, we sampled and sequenced the entire mitochondrial genomes(mitogenomes) of two Tamandua species(Tamandua tetradactyla and T. mexicana) (n=74)from Central and South America, as well as Myrmecophaga tridactyla (n=41) from South America. Within Tamandua, we detected three different haplogroups. The oldest (THI) contained many specimens with the T. tetradactyla morphotype(but also several with the T. mexicana morphotype)and originated in southeastern South America(currently Uruguay) before moving towards northern South America, where the THII haplogroup originated. THII primarily contained specimens with the T. mexicana morphotype (but also several with the T. tetradactyla morphotype) and was distributed in Central America, Colombia, and Ecuador. THI and THII yielded a genetic distance of 4%. THII originated in either northern South America or “in situ” in Central America with haplogroup THIII, which consisted of ~50% T. mexicana and 50% T.tetradactyla phenotypes. THIII was mostly located in the same areas as THII, i.e., Central America,Ecuador, and Colombia, though mainly in the latter.The three haplogroups overlapped in Colombia and Ecuador. Thus, T. tetradactyla and T. mexicana were not reciprocally monophyletic. For this reason, we considered that a unique species of Tamandua likely exists, i.e., T. tetradactyla. In contrast to Tamandua,M. tridactyla did not show different morphotypes throughout its geographical range in the Neotropics.However, two very divergent genetic haplogroups(MHI and MHII), with a genetic distance of ~10%,were detected. The basal haplogroup, MHI,originated in northwestern South America, whereas the more geographically derived haplogroup, MHII,overlapped with MHI, but also expanded into central and southern South America. Thus, Tamandua migrated from south to north whereas Myrmecophaga migrated from north to south. Our results also showed that temporal mitochondrial diversification for Tamandua began during the Late Pliocene and Upper Pleistocene, but for Myrmecophaga began during the Late Miocene.Furthermore, both taxa showed elevated levels of mitochondrial genetic diversity. Tamandua showed more evidence of female population expansion than Myrmecophaga. Tamandua experienced population expansion ~0.6-0.17 million years ago (Mya),whereas Myrmecophaga showed possible population expansion ~0.3-0.2 Mya. However, both taxa experienced a conspicuous female decline in the last 10 000-20 000 years. Our results also showed little spatial genetic structure for both taxa. However,several analyses revealed higher spatial structure in Tamandua than in Myrmecophaga. Therefore,Tamandua and Myrmecophaga were not subjected to the same biogeographical, geological, or climatological events in shaping their genetic structures.
Keywords: Anteaters; Genetic diversity;Mitogenomes; Myrmecophaga; Neotropics;Phylogeography; Spatial structure; Tamandua
INTRODUCTION
The Xenarthra superorder currently represents 31 extant species divided into two orders: i.e., Cingulata and Pilosa.Cingulata contains the armadillos, while Pilosa is further divided into Vermilingua (anteaters) and Folivora (sloths).Xenarthrans are considered the oldest of the placental mammals (O’Leary et al., 2013), and certainly one of the oldest placental lineages (Morgan et al., 2013; Romiguier et al., 2013). Their origin can be traced as far back as 59-65 million years ago (Mya) in South America (Gibb et al., 2016),when they diversified during continental isolation, resulting in over 218 genera (McKenna & Bell, 1997; Möller-Krull et al.,2007). Xenarthrans were highly active during the Great American Biotic Interchange (GABI), with many taxa successfully dispersing from South America into Central and North America (McDonald et al., 2008; Patterson & Pascual,1968). However, as with many other mammalian groups, most xenarthrans became extinct by the end of the Pleistocene,with only a limited number of species surviving, especially during the terminal Pleistocene extinctions that occurred~11 000 years ago. This major extinction event preferentially affected the largest terrestrial forms, such as the giant ground sloths and glyptodonts (Lyons et al., 2004). Extant xenarthrans, such as giant and lesser anteaters, can be regarded as unique examples of the oldest South American endemic radiation of placental mammals (Delsuc et al., 2004,2012; Gibb et al., 2016). This group represents an ideal model for understanding the biogeographical patterns and diversification processes of South America’s “splendid isolation”, following the terminology of Simpson (1980).
Despite the highly specialized and distinct morphologies of the three lineages, the monophyly of the order Xenarthra is classically recognized from morphological (McKenna & Bell,1997) and molecular (Barros et al., 2003, 2008; de melo Casali et al., 2020; Delsuc et al., 2001, 2002, 2004, 2012;Gibb et al., 2016) points of view. All living and fossil xenarthrans exhibit dental reduction, with anteaters at the extreme end with no teeth (Carrol, 1988). One exclusive morphological synapomorphy of Xenarthra is the presence of“xenarthry”, i.e., additional atypical articulations between vertebrae (Engelmann, 1985; Gaudin, 1999; Patterson et al.,1992; Rose & Emry, 1993). Molecularly speaking, xenarthran monophyly is strongly supported by a three consecutive amino acid deletion in the protein αA-crystallin within the lens of the eye. This is unique among studied eutherian mammals (de Jong et al., 1985; Van Dijk et al., 1999).
Delsuc et al. (2001) placed xenarthran radiation—corresponding to the split between Cingulata and Pilosa—around 63 Mya (range: 59-76 Mya) at the boundary of the Cretaceous/Tertiary and dated the split between Vermilingua and Folivora to ~54 Mya in the Early Eocene(range: 51-65 Mya), as verified in later studies (de melo Casali et al., 2020; Delsuc et al., 2004, 2012; Gibb et al.,2016).
New World anteaters (Vermilingua) show a characteristic set of morphological adaptations of the skull, mandible,masticatory myology, digestive organs, and forelimbs, related to myrmecophagy (McDonald et al., 2008; Reiss, 1997;Taylor, 1985). Molecular studies (de melo Casali et al., 2020;Delsuc et al., 2001, 2002, 2003, 2004, 2012; Gibb et al., 2016)support a close relationship betweenMyrmecophaga(Linnaeus, 1758; giant anteater) andTamandua(Linnaeus,1758; lesser anteater)—currently Myrmecophagidae—withCyclopes(Linnaeus, 1758; silky anteater), which belongs to another family (Cyclopedidae). This relationship has been further corroborated by myological (Reiss, 1997) and morphological characters (Gaudin & Branham, 1998). Delsuc et al. (2004) estimated the molecular divergence time within Vermilingua (split between Myrmecophagidae and Cyclopedidae) to have occurred around 40.0 Mya (range:31.8-49.0 Mya), as confirmed in other studies (de melo Casali et al., 2020; Delsuc et al., 2012; Gibb et al., 2016). Thus, the split between anteater lineages seems to have occurred during the main uplift of the Andes around 43 Mya (Marshall &Sempere, 1991).
In the current study, we compared and analyzed the mitochondrial genomes (mitogenomes) of several extant species, namelyTamandua tetradactyla(Linnaeus, 1 758),Tamandua mexicana(Saussure, 1860), andMyrmecophaga tridactyla(Linnaeus, 1 758). The lesser anteater or southern tamandua (T. tetradactyla) is a medium-sized and primarily arboreal mammal. This species is distributed in northern,central, and central-southern South America east of the Andes within a diverse array of habitats, including Chaco, open grassland savanna-like areas (such as Cerrado), wetlands(such as the Pantanal), and mountain tropical regions. It also inhabits transitional forests at elevations up to 2 000 m a.s.l.(Gardner, 2008), but prefers forested areas. Gardner (2008)considered four subspecies ofT. tetradactyla: (1)T.tetradactyla nigra(Geoffroy Saint-Hilaire, 1803). Type locality:French Guiana (Cabrera, 1958). (2)T. tetradactyla quichua(Thomas, 1927). Type locality: Yurac Yacu, San Martín Department, Peru. (3)T. tetradactyla straminea(Cope, 1889).Type locality: Sao Joao (Rio Grande do Sul, Brazil) or Chapada (Mato Grosso; Brazil). (4)T. tetradactyla tetradactyla(Linnaeus, 1 758). Type locality: America meridionali,restricted to Pernambuco, Brazil, by Thomas (1911).
There are several differences between the southern and northern tamandua (T. mexicana) species. Notably,T.mexicanaanteaters can be distinguished by the presence of a black “back-to-front vest” across their backs against a paleyellow background, whereasT. tetradactylaanteaters generally possess uniform golden, brown, black, or partially black-vested fur (Navarrete & Ortega, 2011).Tamandua mexicanais distributed from southern Mexico through Central America to the western Andes (Colombia, Ecuador, and Peru)in South America. It has also been reported from the northern portion of French Guiana (Voss et al., 2001), but not the intermediary northeast South American area (eastern Venezuela, Suriname, and Guyana). This species has been documented in a variety of habitats, including evergreen,deciduous tropical forests, mangroves, secondary forests,savannahs, gallery forests, montane rainforests, and transformed areas (Cuarón, 2005). Four subspecies ofT.mexicanaare recognized (Gardner, 2008; Navarrete &Ortega, 2011): (1)T. mexicana instabilis(Allen, 1904). Type locality: Bonda, Magdalena, Colombia. This subspecies occurs in the western part of Venezuela to the northern Colombian border. (2)T. mexicana mexicana(Saussure,1860). Type locality: Tabasco, Mexico. This subspecies is distributed along the Pacific coast of Mexico, Gulf of Mexico,and from the Yucatan Peninsula to Honduras. (3)T. mexicana opistholeuca(Gray, 1873). Type locality: Colombia (New Grenada). This subspecies ranges throughout Central America to almost the entirety of Colombia. (4)T. mexicana punensis(Allen, 1916). Type locality: Puna Island, Guayas,Ecuador. This subspecies is located along the west coast of Ecuador and in Peru.
The giant anteater (M. tridactyla) is the largest of all anteater species, reaching approximately 30 kg in weight and 180 cm in length. It occupies a variety of habitats, such as rainforests, dry forests, wetlands, and open fields (Aguiar,2004). Unlike other anteaters, this species is entirely terrestrial. Giant anteaters are also highly solitary, and male aggression is common. Historically, they were distributed from Honduras in Central America to the Gran Chaco region of Bolivia, Paraguay, and Argentina, and southern Pampas of Uruguay and Brazil in South America. Gardner (2008) and Gaudin et al. (2018) recognized three extant subspecies:(1)M. tridactyla artata(Osgood, 1912). Type locality:Empalado Savannas, 30 miles east of Maracaibo, Zulia State,Venezuela. This subspecies extends into northeastern Colombia and northwestern Venezuela north and west of the Mérida Andes. (2)M. tridactyla centralis(Lyon, 1906). Type locality: Pacuare, Limón, Costa Rica, with geographical distribution in Central America (where records are few(Genoways & Timm, 2003)) and in northwestern Colombia and northern Ecuador. (3)M. tridactyla tridactyla(Linnaeus,1758). Type locality: America meridionali, but restricted to Pernambuco, Brazil (Thomas 1911). This subspecies occurs in the rest of the South American range east of the Andes from Colombia, Venezuela, and the Guianas south to northern Argentina.
Studies on the phylogeography and population genetics of anteaters remain scarce. At present, only two such studies have been reported forT. tetradactyla(Clozato, 2014; Clozato et al., 2015). Clozato (2014) analyzed eight microsatellites in 176T. tetradactylaspecimens from five Brazilian biomes and found moderate microsatellite genetic diversity and low structure among the Brazilian populations. In addition, given its highest distinctiveness and genetic diversity, the Brazilian Amazon biome was determined to be the diversification center for the species, whereas the populations in the Brazilian Atlantic Forest demonstrated isolation and the other central Brazilian biomes showed no structure. Clozato et al. (2015)also characterized MHC Class II DRB exon 2 diversity and examined spatial distribution across five Brazilian biomes to determine whether different MHC allelic compositions were specific to geographic regions, which could be indicative of local adaptation to differential pathogen pools in the landscape. Their results indicated that the Amazon and Atlantic forests displayed the highest diversity based on number of private alleles and allelic richness, and therefore concluded the existence of MHC allele selection in the different Brazilian biomes.
ForM. tridactyla, Collevatti et al. (2007) reported on the genetic structure, relatedness, and mating strategy of a population in the Emas National Park, Brazil, based on variability at five microsatellite loci, which all displayed low levels of polymorphism. Clozato et al. (2017) also analyzed 77 individuals from seven populations in four Brazilian biomes and sequenced two mitochondrial (mt) markers (control region and cytb) and two nuclear markers (AMELYandRAG2). They found high genetic diversity within several of the populations with signs of population expansion and found significant population differentiation between the Cerrado and Pantanal populations with those from the Amazon.
Herein, we analyzed the complete mitogenomes of the above three species to compare their phylogeography.Mitogenomes are interesting as they can show rapid accumulation of mutations, rapid coalescence time, lack of introns, high number of copies per cell, negligible recombination rate, and haploid inheritance (Avise et al.,1987). Despite representing a single linked locus, selection pressures and evolutionary rates are highly heterogeneous across mitochondrial DNA (mtDNA) (Galtier et al., 2006;Nabholz et al., 2013). For these reasons, mitogenomes are more precise in reconstructing divergence history among closely related species, or within species, than other molecular markers (Moore, 1995).
Thus, we aimed to: (1) Determine the number of taxa, or species, withinTamanduaandMyrmecophagausing mitogenomics; (2) Estimate the time splits within the mitochondrial lineages ofTamanduaandMyrmecophaga;(3) Estimate the mitogenome genetic diversity levels within the different taxa detected withinTamanduaandMyrmecophaga;(4) Determine possible demographic changes within the different taxa detected withinTamanduaandMyrmecophaga;and (5) Describe the comparative spatial genetic structures in bothTamanduaandMyrmecophaga.
MATERIALS AND METHODS
Samples
Samples of hair, muscle, and skins were obtained from 74 specimens ofTamanduasp. and 41 specimens ofM.tridactyla. ForTamandua, 10 Latin American countries were sampled, including Guatemala (n=4), Honduras (n=1),Panama (n=1), Colombia (n=34), Ecuador (n=11), Peru (n=2),Bolivia (n=11), Paraguay (n=2), Argentina (n=6), and Uruguay(n=2). ForMyrmecophaga, six South American countries were sampled, including Colombia (n=20), Ecuador (n=1), Peru(n=9), Bolivia (n=4), Paraguay (n=1), and Argentina (n=6)(Table 1; Figure 1). All samples were from wild specimens(roadkill or specimens collected/hunted by colonists and indigenous people) except for one specimen ofTamanduasp.from Rosario Zoo (Argentina).

Table 1 Sources of Tamandua sp. (n=74) and Myrmecophaga tridactyla (n=41) specimens collected and analyzed(mitogenomes) from Latin America and South America,respectively

Continued
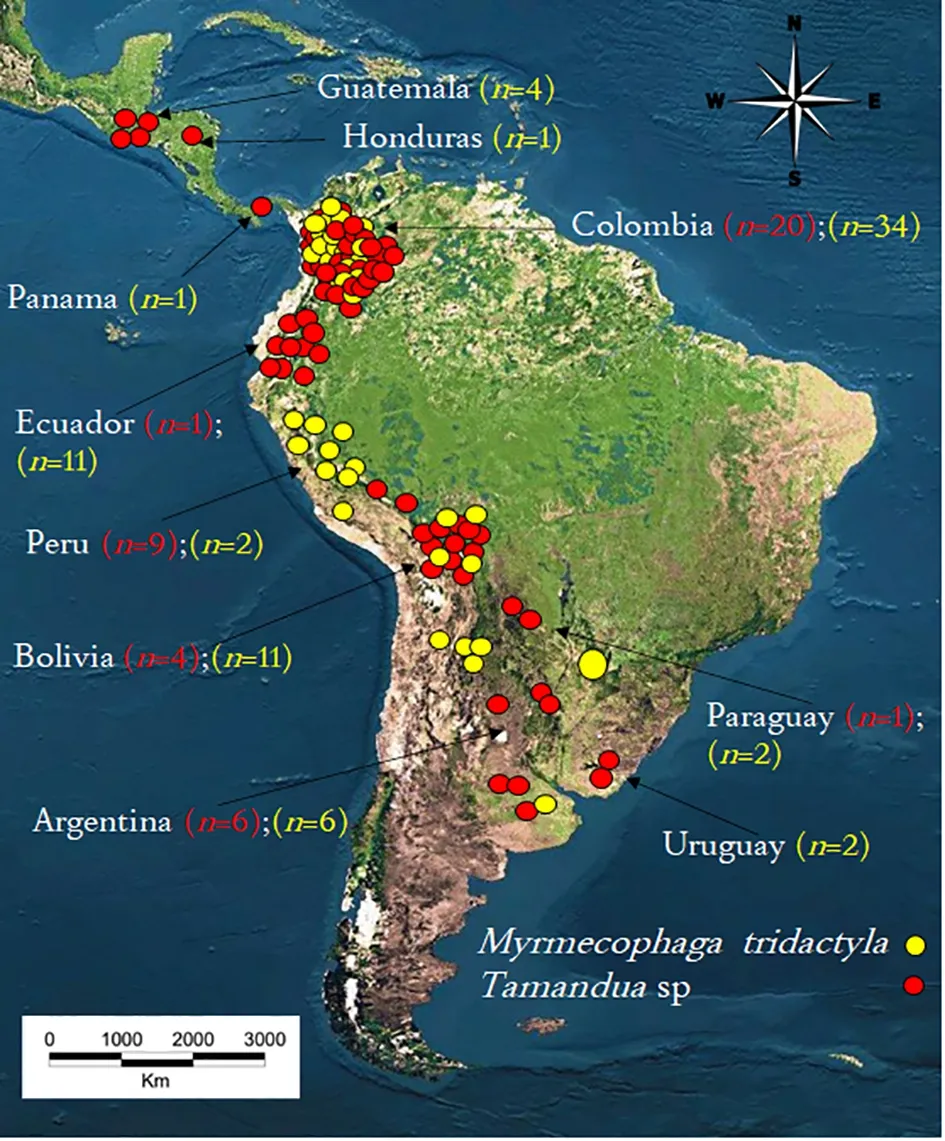
Figure 1 Map of Latin America identifying sampling locations of Tamandua and Myrmecophaga
Molecular methods
DNA was extracted and isolated from the hair, muscle, and skin samples using the QIAamp DNA Micro Kit (Qiagen,Netherlands) following the protocols provided by the manufacturer. Muscle extraction followed the “DNA Purification from Tissues” protocol. Mitogenomes were sequenced by long-template polymerase chain reaction(PCR), which minimizes the chance of amplifying mitochondrial pseudogenes from the nuclear genome (numts)(Raaum et al., 2005; Thalmann et al., 2004). Four sets of primers (MYR1R-1F, MYR2R-2F, MYR3R-3F, and MYR4R-4F) were used to generate overlapping amplicons from 3 511 to 4 123 bp in length, thereby enabling a quality test for genome circularity (Bensasson et al., 2001; Thalmann et al.,2004). PCR amplification of mtDNA was carried out using a Long-Range PCR Kit (Qiagen, Netherlands), with a reaction volume of 25 μL and a reaction mix consisting of 2.5 μL of 10×Long-Range PCR Buffer, 500 μmol/L of each deoxyribonucleoside triphosphate (dNTP), 0.6 μmol/L of each primer, 1 unit of Long-Range PCR Enzyme, and 50-250 ng of template DNA. Cycling conditions were as follows: 94 °C for 5 min, followed by 45 cycles of denaturing at 94 °C for 30 s,primer annealing at 50-57 °C (depending on primer set) for 30 s, and an extension at 72 °C for 8 min, followed by 30 cycles of denaturing at 93 °C for 30 s, annealing at 45-52 °C(depending on primer set) for 30 s, and extension at 72 °C for 5 min, with a final extension at 72 °C for 8 min.
Both mtDNA strands were sequenced directly using BigDye Terminator v3.1 (Applied Biosystems, USA). Sequencing products were analyzed on an ABI 3730 DNA Analyzer system (Applied Biosystems, USA). The circular mtDNA was 16 543 bp long, and contained 13 protein-coding genes, two ribosomal RNA (rRNA) genes, 22 transfer RNA (tRNA) genes,and the control region of 1 100 bp. All protein-coding genes(exceptND6), two rRNA genes, and 14 tRNA genes were on the heavy strand. The mitogenome was annotated using MITOS software (Bernt et al., 2013) and verified with ExPASy software (Gasteiger et al., 2003). The protein-coding genes commonly had an ATG start codon (10 out of 13) and a TAA stop codon (eight out of 13).
Sequences were assembled and edited using Sequencher v4.7 (Gene Codes Corporation, USA). Overlapping regions were examined for irregularities such as frameshift mutations and premature stop codons. A lack of such irregularities indicates an absence of contaminating numt sequences.
All gene alignments (16 543 bp) were concatenated after removing problematic regions using Gblocks v0.91 (Talavera& Castresana, 2007) under a relaxed approach. This software removes all poorly aligned regions and is effective in phylogenetic studies with highly divergent sequences(Castresana, 2000; Talavera & Castresana, 2007). The best partition and most suitable evolutionary models were defined with PartitionFinder v.2.1 (Lanfear et al., 2012) using Bayesian Information Criterion (BIC) (Minin et al., 2003), assuming“greedy” as the search algorithm and “linked” as the branch length. This program was used to determine the optimal model of evolution and partitioning scheme simultaneously (for partitions, codons 1+2 combined, and codon 3, for each gene,including the control region and RNAs). Afterwards, we generated a concatenated matrix using SequenceMatrix v1.7.6 (Vaidya et al., 2011).
The GenBank accession numbers of the analyzed anteater specimens are MW465709-MW465748 and MW489364-MW489421.
Phylogenetic, genetic heterogeneity, and genetic diversity analyses
jModelTest v2.0 (Darriba et al., 2012), Kakusan4 (Tanabe,2011), and MEGA X v10.0.5 (Kumar et al., 2018) were applied to determine the best evolutionary mutation model for the sequences analyzed for each individual gene, different partitions, and all concatenated sequences. BIC (Schwarz,1978) was used to determine the best evolutionary nucleotide model for the anteater mitogenomes.
Phylogenetic trees were constructed using maximumlikelihood (ML) procedures. The ML tree was obtained using RAxML v8.2.X (Stamatakis, 2014) implemented in the CIPRES Science Gateway (Miller et al., 2010). The HKY+G+I model (Hasegawa, Kishino, and Yano model+gamma distributed rate variation among sites+proportion of invariable sites; Hasegawa et al., 1985) was used to search the ML tree.We estimated node support using the rapid-bootstrapping algorithm (−f a -x option) for 1 000 non-parametric bootstrap replicates (Stamatakis et al., 2008). The haplogroups of anteaters were considered significant when bootstraps were higher than 80% (lax limit; Hillis & Bull, 1993).
BEAST v2.5.1 was run to estimate the time to the most recent common ancestor (TMRCA) for the different nodes of the Bayesian inference (BI) trees. We used a prior of 12.5±0.5 Mya for the split of the ancestor ofTamandua-Myrmecophaga(average values from de melo Casali et al.,2020; Delsuc et al., 2004, 2012; Gibb et al., 2016).
To reconstruct the possible relationships among theTamanduaandMyrmecophagahaplotypes analyzed and to estimate the possible divergence times among these haplotypes, we constructed a median-joining network (MJN)using Network v4.6.0.1 (Fluxus Technology Ltd., Colchester,Essex, England) (Bandelt et al., 1999). Additionally, the ρ statistic (Morral et al., 1994) and its standard deviation(Saillard et al., 2000) were estimated and transformed into years. The ρ statistic is unbiased and highly independent of past demographic events; furthermore, this “borrowed molecular clocks” approach uses direct nucleotide substitution rates inferred from other taxa (Pennington & Dick, 2010).Here, we used an evolutionary rate of 2.75% per one million years (Horai et al., 1995; Nabholz et al., 2008), representing one mutation each 2 198 years for the 16 543 bp analyzed.Network analyses can be more useful in the reconstruction of evolutionary history within a species or among closely related species than bifurcating trees, as is the case for the tamandua and giant anteaters analyzed.
TheHST,KST,KST*,γST,NST, andFSTstatistical procedures(Hudson et al., 1992) were applied to determine the overall genetic heterogeneity among the different taxa withinTamanduaandMyrmecophagadetected during phylogenetic analyses. We obtained indirect gene flow estimates for all taxa, assuming an infinite island model (Wright, 1965).Significance was estimated with permutation tests using 10 000 replicates. Genetic heterogeneity and gene flow statistics were calculated by taxon pairs. We usedFSTtests with Markov chains, 10 000 dememorization parameters, 20 batches, and 5 000 iterations per batch. All statistics were calculated using DNAsp v5.1 (Librado & Rozas, 2009) and Arlequin v3.5.1.2 (Excoffier& Lischer, 2010).
The Kimura 2-parameter (2P) genetic distance (Kimura,1980) was applied to determine the percentage of genetic differences among the haplogroups detected in bothTamanduaandMyrmecophaga. The Kimura 2P genetic distance is a standard measurement for barcoding tasks(Hebert et al., 2003, 2004). Kartavtsev (2011) analyzed mtCOIsequences from 20 731 vertebrate and invertebrate species and obtained 0.89%±0.16% for populations within species,3.78%±1.18% for subspecies or semi-species, and 11.06%±0.53% for species within a genus. For mtCOII,Ascunce et al. (2003) and Ruiz-García et al. (2014) reported an average genetic distance of ~8% among species within a genus and ~2%-5% for subspecies. For cytb, Bradley &Baker (2001) and Baker & Bradley (2006) stated that <2%represented equal intra-specific variation, 2%-11% required additional study, and >11%-13% indicated specific recognition. Therefore, for mitochondrial genes, we considered an average of >4%-10% for possible subspecies,>12%-13% for different species within the same genus, and>16%-18% for species in different genera (Kartavtsev, 2011).
We used the following statistics to determine the genetic diversity of theTamanduaandMyrmecophagasamples, and for the main haplogroups within them: number of haplotypes,haplotype diversity (Hd), nucleotide diversity (π), andθstatistics by sequence. These genetic diversity statistics were calculated using DNAsp v5.1 (Librado & Rozas, 2009).
Demographic changes
We relied on three procedures to detect possible historical population changes in theTamanduaandMyrmecophagasamples, and in the haplogroups detected within them. (1) We used the Fu and Li’sD* andF* tests (Fu & Li, 1993), Fu’sFSstatistic (Fu, 1997), Tajima’sDtest (Tajima, 1989), andR2statistic (Ramos-Onsins & Rozas, 2002). Both 95%confidence intervals and probabilities were obtained with 10 000 coalescence permutations. (2) Mismatch distribution(pairwise sequence differences) was obtained following Rogers & Harpending (1992) and Rogers et al. (1996). We used raggednessrgto determine the similarity between the observed and theoretical curves. To estimate the time when a demographic change began, we used a generation time of one year forTamanduaand three years forMyrmecophaga(Rojano et al., 2014). These demographic analyses were carried out using DNAsp v5.1 (Librado & Rozas, 2009) and Arlequin v3.5.1.2 (Excoffier & Lischer, 2010). (3) A Bayesian skyline plot (BSP) was obtained using BEAST v2.5.1(Bouckaert et al., 2014; Drummond et al., 2012) and Tracer v.1.7 (Rambaut et al., 2018). The coalescent-Bayesian skyline option in the tree priors was selected with four steps and a piecewise-constant skyline model with 30 million generations(first six million discarded as burn-in), kappa with log-normal(1, 1.25), and skyline population size with uniform (0, infinite;initial value 80). Marginal densities of temporal splits were analyzed, and the Bayesian skyline reconstruction option was selected for the trees log file using Tracer v1.7. We selected a stepwise (constant) Bayesian skyline variant with maximum time as the upper 95% high posterior density (HPD) and trace of the root height as treeModel.rootHeight. We considered~0.6-2.5 Mya forTamanduaand ~2-4 Mya forMyrmecophagafor analysis. Nevertheless, all these demographic procedures have several caveats. Selection can affect effective population size, reducing the effective number for a time and increasing the coalescence rate later (Schrider et al., 2016). The same occurs with small changes in the mutation rate (μ), which can greatly affect the effective number and, in turn, estimated divergence time (Sheehan et al., 2013).
Spatial genetic analyses
For the following analyses, we used the geographic coordinates of the specimens without classifying them into populations, except for the autocorrelation index for DNA analysis (AIDA procedure, where we used populations (or determined regional areas with their average geographic coordinates). Ten populations were used forTamanduaand eight populations were used forMyrmecophaga. Thus, we analyzed whether specimen or population analysis could detect spatial genetic structure.
Mantel test (Mantel, 1967) was used to detect possible relationships between a genetic matrix ofTamanduaspecimens and ofMyrmecophagaspecimens (Kimura 2P genetic distance) and the geographic distance matrix among specimens analyzed for each taxon. In this study, Mantel’s statistic was normalized according to Smouse et al. (1986),which transforms the statistic into a correlation coefficient.
The spatial autocorrelation analysis utilized the Ay statistic(Miller, 2005) for each distance class (DC), i.e.,Ay=Σi=1, n Σj>i, n (Dijwyij)/Σi=1, n Σj>i, nwyij, wherenis the number of individuals in the dataset andDijis the genetic distance between observationsiandj. The elements of the binary matrix,Wyij, take on values of 1 if the geographical distance between observationsiandjfall within the boundaries specified for a specific DC, or are 0 otherwise. Ay can be interpreted as the average genetic distance between pairs of individuals that fall within a specific DC, with a value of 0 when all individuals within a DC are genetically identical and a value of 1 when all individuals within a DC are completely dissimilar.The probability for each DC was obtained using 1 000 randomizations. For this analysis, which was carried out with AIS software (Miller, 2005), there were 10 defined DCs for bothTamanduaandMyrmecophaga(Tamandua: 0-197 km;197-334 km; 334-514 km; 514-721 km; 721-1 023 km;1 023-1 258 km; 1 258-1 484 km; 1 484-2 128 km; 2 128-2 557 km; 2 557-4 025 km;Myrmecophaga: 0-127 km;127-245 km; 245-383 km; 383-490 km; 490-641 km;641-966 km; 966-1 218 km; 1 218-1 447 km; 1 447-1 967 km;1 967-2 602 km).
We used AIDA as a second spatial autocorrelation procedure (Bertorelle & Barbujani, 1995) for bothTamanduaandMyrmecophagawith theIIandccstatistics. To connect these geographic localities within each distance class, we used the Gabriel-Sokal network (Gabriel & Sokal, 1969; Ruiz-García, 1993, 1994, 1997, 1999; Matula & Sokal, 1980) and Delaunay triangulation with elimination of the crossing edges(Isaaks & Srivastava, 1989; Ripley, 1981; Upton & Fingleton,1985). The Bonferroni (Oden, 1984) and Kooijman tests were applied to determine the significance of the autocorrelation coefficients. Two AIDA runs, one forTamanduaand one forMyrmecophaga, were carried out with five DCs (Tamandua: 1 DC: 0-943 km; 2 DC: 943-1 897 km; 3 DC: 1 897-2 414 km;4 DC: 2 414-4 082 km; 5 DC: 4 082-7 001 km;Myrmecophaga: 1 DC: 0-1 066 km; 2 DC: 1 066-1 577 km; 3 DC: 1 577-2 683 km; 4 DC: 2 683-3 790 km; 5 DC: 3 790-4 501 km).
Further spatial analysis was carried out using Monmonier’s maximum difference algorithm (MMDA) (Monmonier, 1973)with AIS software (Miller, 2005). This geographical regionalization procedure can be used to detect the locations of putative barriers to gene flow by iteratively identifying sets of contiguous, large genetic distances along connectivity networks (Dupanloup et al., 2002; Manel et al., 2003; Manni et al., 2004). Delaunay triangulation (Brouns et al., 2003;Watson, 1992) was used to generate the connectivity network among sampling points. In this procedure, a graphical representation of putative “barriers” inferred by the algorithm is superimposed over the connectivity network to detect rapid identification of important geographical features reflected by the genetic dataset. Here, we used this procedure to detect the three most potentially important geographical barriers contained in the dataset forTamanduaandMyrmecophaga.
RESULTS
Phylogeographic patterns
The best nucleotide substitution models were HKY+G+I (-Ln=99 224.03) forTamanduaand HKY+G+I (-Ln=61 638.17) forMyrmecophaga. Thus, the best nucleotide substitution model was the same for both genera.
The ML tree forTamandua(Figure 2) showed that the first two divergentTamanduaspecimens were both from southern South America (Uruguay). A clade (bootstrap support (BS):88%) with 30 specimens appeared later. A considerable fraction of these specimens (63%) showed theT. tetradactylaphenotype and were sampled in southern or central South America (Argentina, Paraguay, Bolivia, and southern Peru).However, some specimens were also from northern South America (Colombia and Ecuador; 37%) and both trans-Andean and cis-AndeanTamanduaspecimens were included.In fact, two trans-Andean ColombianTamanduaspecimens(one from Magdalena Valley and one from Tayrona NP, Santa Marta, Caribbean coast) and one trans-Andean Ecuadorian specimen (Santo Domingo de Tsáchilas) with theT. mexicanaphenotype were in this clade, which was named THI(Haplogroup I). The following divergent clade (Haplogroup II,THII) (BS=94%) was composed of 17 specimens. Most of these specimens showed theT. mexicanaphenotype (82%)and were from Central America (Panama, Honduras, and Guatemala) and Colombian and Ecuadorian trans-Andean areas (Colombia: Caquetá, Córdoba, Nariño, Tolima, and Valle del Cauca departments; Ecuador: Santo Domingo de Tsáchilas Province), coinciding with the distribution ofT.mexicana. Nevertheless, three specimens (18%) presented theT. tetradactylaphenotype, two from the Tolima Department in Colombia and one from the Cochabamba Department in Bolivia. Therefore, as for THI, this haplogroup was also comprised of mixed specimens of the two putativeTamanduaspecies. The third clade (Haplogroup III, THIII)(BS=99%) was comprised of 25 specimens, with a strong mix of bothT. mexicana(52%) andT. tetradactyla(48%). SomeT.mexicanaspecimens were from Central America (Guatemala)and Ecuadorian trans-Andean areas (Manabí and Pichincha Provinces), but most were from the trans-Andean area of Colombia (Antioquia, Atlántico, Chocó, Magdalena, Risaralda,Tolima, and Valle del Cauca departments). TheT. tetradactylaphenotype specimens were primarily from the Colombian Eastern Llanos region (Arauca, Meta, and Vichada departments), and Colombian (Caquetá Department) and Ecuadorian Amazon (Napo Province). One unique exception was theT. tetradactylaphenotype specimen from Rosario(Argentina). However, this individual was from a zoo with unknown origin, and thus may have originated from northern South America.
The ML tree forMyrmecophaga(Figure 3) showed two haplogroups with some internal heterogeneities. The first(BS=99%; MHI) was comprised of 10 specimens. The most divergent specimen was from the Peruvian Amazon (Masisea,Ucayali River), while the remaining specimens were from diverse areas in northern South America, including the Peruvian Ucayali River, Peruvian San Martin Department,Bolivian Santa Cruz Department, Colombian Amazon Department, and Colombian Eastern Llanos region (Meta and Casanare departments). The second major clade (BS=99%)was haplogroup II (MHII), which consisted of 31 specimens.The most divergent specimen within this clade was from the Colombian Eastern Llanos region (Meta Department). Within this haplogroup, there were three other clusters with high bootstrap values but with no geographical pattern. The first(BS=99%) contained one specimen from Paraguay; the second (BS=97%) consisted of specimens from Argentina,Bolivia, Colombia, and Peru; and the third (BS=98%)contained specimens from Argentina, Bolivia, Colombia(different departments with different biomes), Ecuador, and Peru. As mentioned above, no spatial structure was observed within these clusters, except for a potential small cluster with three specimens from the Tucuman Province in Argentina.

Figure 2 Maximum-likelihood tree of 74 Tamandua sp. specimens based on entire mitogenomes
The MJN forTamandua(Figure 4) showed that the first haplotypes to appear, i.e., basal haplotypes (less differentiated from the outgroupMyrmecophaga), were H45 and H46, which corresponded to the two analyzed Uruguayan specimens with theT. tetradactylaphenotype. These results support an origin for the current haplotypes ofTamanduain southern and eastern South America. These haplotypes are the origins of the THI haplotypes (e.g., H27, H40, H41, H43,H44), which were mainly distributed in southern and central South America (Argentina, Paraguay, Bolivia, and southern Peru) with theT. tetradactylaphenotype. However, some haplotypes in this group arrived in northern South America(Ecuador and Colombia) and exhibited theT. mexicanaphenotype. THI later generated the haplotypes of THII formed by specimens in northern South America (Colombia and some from Ecuador) and Central America (Panama, Honduras, and Guatemala). Most specimens exhibited theT. mexicanaphenotype, although several from the inter-Andean valleys of Colombia (Tolima Department) and one from the Cochabamba Department (Bolivia) showed theT. tetradactylaphenotype. THII generated H69 and H34, with the latter generating all the other haplotypes of THIII, which was composed of both theT. mexicana(Colombia, Ecuador, and Central America) andT. tetradactylaphenotypes (Colombian Eastern Llanos region, with some haplotypes also distributed in Colombian and Ecuadorian Amazon).

Figure 3 Maximum-likelihood tree of 41 Myrmecophaga tridactyla specimens based on entire mitogenomes
The MJN forMyrmecophaga(Figure 5) showed two welldelimited groups, similar to the phylogenetic trees. The most basal haplotypes were those from MHI (H7, H8, H11, H15,H20, H22, H23, and H28), which were most related to the outgroupTamandua. In contrast to the last genus, these haplotypes were generated in northwestern South America,approximately within the current Peruvian and Colombian Amazon or current Colombian Eastern Llanos region.Radiation of haplotypes began in the Colombian Eastern Llanos (H28). In turn, haplotypes of MHII were generated,which later colonized central and southern areas of South America (Bolivia, Paraguay, and Argentina). They also remained in sympatry in the Colombian Eastern Llanos and western Amazon regions, where MHI originated.
Thus, the geographical origin forTamanduawas in southern South America and colonization was from south to north,whereas the geographical origin forMyrmecophagawas in northwestern South America and colonization was from north to south.
Temporal splits within Tamandua and Myrmecophaga
ForTamandua, using the MJN procedure, we estimated that divergence between the most divergent and basal Uruguayan haplotypes to the first haplotype (H69) of THIII (most derived)occurred 1 631 750±40 164 years ago. If we included the most derived haplotypes within THIII, temporal divergence increased to 3 468 583±32 069 years ago. The first estimate correlated with the beginning of the Calabrian Stage(Pleistocene) and the second correlated with the last phase of the Pliocene. Mitochondrial diversification in THI began~878 526±140 451 years ago, in THII began ~583 636±113 781 years ago, and in THIII began ~713 333±93 327 years ago. Hence, internal diversification in the three haplogroups occurred within the Pleistocene. The BI estimate for the beginning of the mitochondrial diversification was~1.8 Mya (95% HPD: 1.39-3.95 Mya), which was very similar to the first estimate obtained via the MJN procedure.Mitochondrial diversification within THI was estimated to have begun ~1.19 Mya (95% HPD: 0.59-1.65 Mya), within THII was estimated to have begun ~0.68 Mya (95% HPD: 0.29-1.37 Mya), and within THIII was estimated to have begun~1.31 Mya (95% HPD: 0.81-2.11 Mya). Therefore, internal haplogroup diversification was slightly earlier than that obtained with MJN but was still within the Pleistocene.
ForMyrmecophaga, the MJN procedure detected a temporal split between MHI and MHII ~2 310 163±42 781 years ago. Internal mitochondrial diversification in MHI was estimated to have occurred ~203 209±41 442 years ago, while internal diversification in MHII was estimated to have occurred~516 435±55 717 years ago. The BI estimates were somewhat earlier than those obtained with the MJN procedure. The haplotype of the Peruvian Masisea specimen(Ucayali River) diverged ~6.12 Mya (95% HPD: 4.59-13.17 Mya). The temporal split between the ancestors of MHI and MHII was estimated to have occurred ~4.98 Mya (95%HPD: 3.39-10.96 Mya), with internal mitochondrial diversification in MHI and MHII estimated to have occurred~1.3 Mya (95% HPD: 1.27-8.22 Mya) and ~3.76 Mya (95%HPD: 2.13-7.65 Mya), respectively. Thus, the BI temporal estimates were earlier than the MJN temporal estimates.

Figure 4 Median-joining network (MJN) with haplotypes from entire mitogenomes of Tamandua sp. sampled in Latin America

Figure 5 Median-joining network (MJN) with haplotypes from entire mitogenomes of Myrmecophaga tridactyla sampled in South America
Genetic heterogeneity and genetic distances
Genetic heterogeneities among the three haplogroups detected inTamanduaas well as between the two haplogroups detected inMyrmecophagawere significant(Table 2). ForTamandua, all statistical analyses were significant (except for χ2) (i.e.,γST=0.581,P=0.003;NST=0.681,P=0.002 1;FST=0.669,P=0.002 4) with low gene flow estimates (Nm=0.36, 0.23, and 0.25, respectively).Between haplogroup pairs, the lowest genetic heterogeneity was between THI and THII (γST=0.262,P=0.04;NST=0.428,P=0.005;FST=0.423,P=0.005), followed by THI and THIII(γST=0.551,P=0.002 8;NST=0.711,P=0.001 9;FST=0.698,P=0.002 1), and THII and THIII (γST=0.568,P=0.004 8;NST=0.743,P=0.000 3;FST=0.732,P=0.000 4). Curiously, no different morphotypes were found forMyrmecophaga, as detected inTamandua, and molecular genetic heterogeneity was even higher. All statistical analyses were significant(γST=0.581,P=0.002;NST=0.796,P=0.000 1;FST=0.788,P=0.000 1) and their respective gene flow estimates were smaller than those obtained for theTamanduahaplogroups(Nm=0.36, 0.13, and 0.13, respectively).
The same occurred for genetic distances. ForTamandua,the Kimura 2P genetic distances between TH1 and TH2, TH1 and TH3, and TH2 and TH3 were 4.0%±0.5%, 10.9%±1.1%,and 10.5%±1.1%, respectively. ForMyrmecophaga, the Kimura 2P genetic distance between MH1 and MH2 was 9.7%±1.1%. Thus, there was similar genetic differentiation between the two haplogroups ofMyrmecophagaand the most differentiated groups ofTamandua. These values were in the high range for subspecies. The genetic distance betweenTamanduaandMyrmecophaga(21%) was typical for different species of different genera.

Table 2 Genetic heterogeneity and gene flow based on mitogenomes of Tamandua sp. and Myrmecophaga tridactyla analyzed in this study
Mitochondrial genetic diversity
The mt genetic diversity statistics forTamanduaandMyrmecophagaare shown in Table 3. Total samples, as well as the three haplogroups, showed elevated genetic diversity levels (total samples:π=0.063 9±0.000 3; THI:π=0.026 2±0.000 2; THII:π=0.017 8±0.002 7; THIII:π=0.035 8±0.000 7).
ForMyrmecophaga, the total samples, as well as the two haplogroups, showed elevated genetic diversity levels (total samples:π=0.050 7±0.000 5; MHI:π=0.014 8±0.000 5; MHII:π=0.024 3±0.000 6). The highly elevated nucleotide diversities(π) for the total samples of both genera were likely due to the existence of the well-differentiated haplogroups within both genera.
Demographic changes
For demographic changes and mismatch distributions(Supplementary Figure S1),Tamanduashowed more evidence of female population expansion thanMyrmecophaga. Overall, four out of six tests showed significant evidence of expansion inTamandua(Fu & LiD*=-2.51,P=0.022; Fu & LiF*=-2.15,P=0.027;FS=-28.44,P=0.000 2; and mismatch distribution,rg=0.000 8,P=0.000 001). Although significant, the mismatch distribution for allTamanduasamples showed two curves because two or more genetically different populations were included in the sample and/or there were two population expansions. THI showed very strong evidence of female population expansion based on all six tests (TajimaD=-1.88,P=0.013; Fu & LiD*=-2.24,P=0.036; Fu & LiF*=-2.51,P=0.026;FS=-16.38,P=0.000 01, R2=0.06,P=0.005; and mismatch distribution,rg=0.006,P=0.009 7). However, THII and THIII showed little evidence of female population expansion (i.e., THII:FS=-7.91,P=0.002, and R2=0.08,P=0.012; and THIII: TajimaD=-1.51,P=0.045; R2=0.07,P=0.016; and mismatch distribution,rg=0.006,P=0.027). THI showed stronger evidence of population expansion as it is the oldest haplogroup detected withinTamandua(Uruguayan specimens excluded), whereas THII and THIII are more recent and derived haplogroups.Additionally, except for THII, three out of four mismatch distribution analyses forTamanduawere significant for female population expansion, allowing us to estimate when the expansion began. Thus, the estimate for allTamanduawas~470 300 years ago, for THI was ~271 000 years ago, and for THIII was ~168 200 years ago.
ForMyrmecophagaoverall, we found no evidence of female population expansion. This may be because the samples were composed of two very different populations. Individually,however, each haplogroup showed evidence of female population expansion. In the case of MHI, three out of six tests were significant (TajimaD=-1.96,P=0.006; Fu & LiD*=-2.28,P=0.01; Fu & LiF*=-2.49,P=0.01), while in the case of MHII,five out of six tests were significant (TajimaD=-1.9,P=0.009;Fu & LiD*=-2.54,P=0.022; Fu & LiF*=-2.75,P=0.02;FS=−5.02,P=0.048, R2=0.065,P=0.014). However, neither of the mismatch distribution analyses were significant, which differs from that found forTamandua, and therefore we did not estimate when female population expansion may have begun forMyrmecophaga.
The BSP analyses (Supplementary Figure S2) showed interesting and complementary results to the previous demographic analyses. Overall,Tamanduashowed a strong female population increase ~250 000-300 000 years ago(similar to that reported for the mismatch distribution), which stopped around 50 000 years ago. For THI, female population expansion was dated to ~200 000-250 000 years ago (very similar to that estimated by mismatch distribution, 271 000 years ago), with a strong decrease in the last 20 000 years.For THIII, female increase began 0.6 Mya (considerably earlier than that obtained with mismatch distribution). Overall,Myrmecophagashowed a female population increase~200 000-250 000 years ago and, more recently, a population decrease in the last 6 000 years and an increase in the last century. For MHI, BSP analysis showed a constant female population, with a strong decrease in the last 10 000-20 000 years, while for MHII, analysis showed a slight increase 200 000 years ago and a very strong decrease in the last 10 000-20 000 years. Therefore, based on BSP analysis, both genera showed a female population increase ~200 000-400 000 years ago, although it was clearer forTamandua, and both demonstrated a female population decrease during the last 10 000-20 000 years.

Table 3 Genetic diversity statistics (±standard deviation) for Tamandua sp. and Myrmecophaga tridactyla
Spatial structure
Mantel tests forTamanduaandMyrmecophagadid not reveal any significant relationship between geographic and genetic distances (Tamandua: r=0.051,P=0.075;Myrmecophaga:r=0.042,P=0.251; Supplementary Figure S3).
Spatial autocorrelation analysis with 10 DCs forTamanduashowed a non-significant overall correlogram (V=0.002 5,P=0.83), although the two most negative values for the last two DCs (9 DC: 2 128-2 557 km; 10 DC: 2 557-4 025 km) did not reach statistical significance (Supplementary Figure S4).Spatial autocorrelation analysis with 10 DCs forMyrmecophagaalso showed a non-significant overall correlogram (V=0.004 7,P=0.80). Only two DCs (127-245 km)showed a significant negative value (P=0.028 8)(Supplementary Figure S4). This suggests the existence of certain genetic patches forMyrmecophagawith a diameter of around 245 km. However, for both taxa, there was no significant spatial autocorrelation structure. The different haplogroups, to a high degree, overlapped and were sympatric in bothTamanduaandMyrmecophaga.Tamanduashowed some isolation-by-distance (negative value of the last DC) but did not support the existence of two well-differentiated species (T. mexicanaandT. tetradactyla) when the overall data were analyzed.
AIDA (Table 4) with populations and five DCs showed several important differences betweenTamanduaandMyrmecophaga. The correlogram forTamanduasupported some influence of isolation-by-distance. Based on a 95%confidence interval and Moran’sIstatistic, the first DC (I=0.040 8;0-943 km) and second DC (I=0.031 8; 943-1 897 km) were significantly positive, while the fourth DC (I=-0.083 3; 2 414-4 082 km) and fifth DC (I=-0.136 9; 4 082-7 001 km) were significantly negative, which agrees relatively well with a certain isolation-by-distance. Based on a 99% confidence interval, only the fifth DC was significantly negative. For Geary’sccoefficient, no DC was significant under 95% or 99% confidence intervals. ForMyrmecophaga, AIDA did not recover any significant DCs using Moran’sIand Geary’scor 95% and 99% confidence intervals. Hence, using populations,not specimens as in the previous analyses, there was certain evidence of isolation-by-distance forTamandua, but no spatial structure forMyrmecophaga.
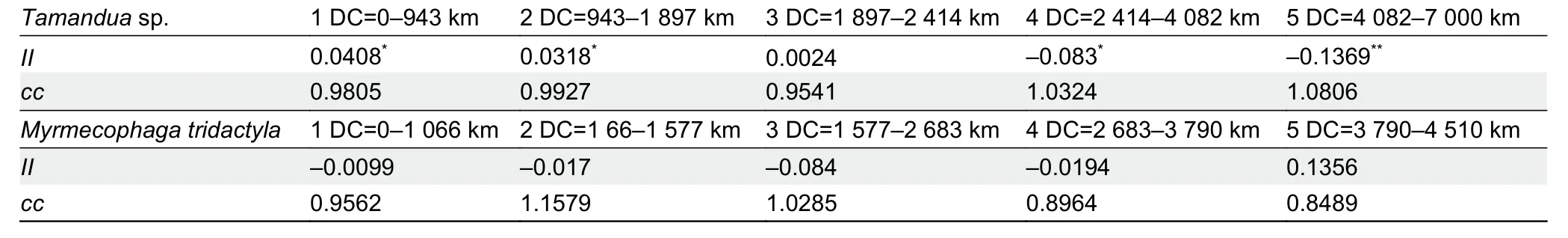
Table 4 Spatial autocorrelation analyses using AIDA with five distance classes (DC), II index, and cc coefficient for Tamandua sp. and Myrmecophaga tridactyla
Finally, we used MMDA analyses (Figure 6) to detect the three most important barriers forTamanduaandMyrmecophaga. ForTamandua, the first barrier (blue)separated the specimens sampled in Central America,Colombia, Ecuador, and some eastern points in Bolivia and Argentina (Misiones Province) from the remaining specimens analyzed; the second barrier (green) isolated some specimens from the Cochabamba Department (Bolivia); and the third barrier (green blue) separated the specimens from Uruguay and some parts of Argentina. ForMyrmecophaga, the first barrier (blue) separated specimens from Peru and part of Bolivia to northern-western Argentina (Jujuy and Tucuman Provinces); the second barrier (green) isolated specimens from northern Argentina, a major part of Bolivia, and Paraguay; and the third barrier (green blue) contained specimens from different Colombian departments (e.g.,Cundinamarca, Meta, and Tolima). Thus, the potential geographic barriers differed for the two genera.
DISCUSSION
How many species are within Tamandua and Myrmecophaga
This is the first mitogenomic study trying to elucidate the number of taxa withinTamanduaandMyrmecophaga. mtDNA has been extremely useful in detecting new or previously overlooked taxa (see Derenko et al., 2012; Krause et al.,2010; Sawyer et al., 2015). Traditionally, two species ofTamanduahave been considered (Gardner, 2008), i.e., one in the trans-Andean area of northern South America and southern Mexico and Central America with a well-developed black vest on its body (T. mexicana), and one distributed in the cis-Andean area of South America without a welldeveloped black vest (T. tetradactyla). Our study demonstrated the existence of three haplogroups. THI was mainly composed ofT. tetradactylaspecimens with a preponderance from central and southern South America but also from northern South America, and even severalT.mexicanaspecimens from the trans-Andean area of Ecuador and Colombia. THII was composed mainly ofT. mexicanaspecimens from Central America and the trans-Andean area of Colombia and Ecuador, but also severalT. tetradactylaspecimens from Colombia and Bolivia. THIII was composed—in similar proportions—ofT. mexicanaandT.tetradactylafrom Colombia, Central America, and Ecuadorian Amazon. Thus, the three detected haplogroups were intermixed with specimens ofT. mexicanaandT. tetradactyla.We did not detect monophyla reciprocity between the twoa prioriputative species ofTamandua. This is a strong indication that only one species should be considered within this genus,i.e.,T. tetradactyla(Linnaeus, 1 758 vs. Saussure, 1860),because the genetic differentiation was a gradual and continuous process likely driven by anagenesis or phyletic evolution, not by cladogenesis speciation (Eldredge & Gould,1972; Gould & Eldredge, 1993). Furthermore,Tamanduashowed no clear spatial genetic structure, although, in some analyses, we detected isolation-by-distance, which usually occurs in gradual transformation processes. Additionally, this shows that the presence or absence of the black vest on the body has no systematic or phylogenetic value forTamandua.This explains the rare distribution of oneT. mexicanaspecimen in French Guiana described by Voss et al. (2001).The black vest is likely controlled by one or a few genes and its presence or absence is influenced by genetic drift or founder events. Similarly, melanism inTamanduahas no taxonomic or phylogenetic importance. For instance, we analyzed two melanistic specimens from Ecuador (Zamora and Morona-Santiago provinces), which were contained in THI along with mixed specimens with the typicalT. tetradactylagolden phenotype and some specimens with theT. mexicanaphenotype. Ríos-Alvear & Cadena-Ortíz (2019) reported six records of melanism inT. tetradactylafrom southern Ecuador,Zamora Chinchipe, and Morona Santiago, the same provinces sampled here (in fact, we sequenced two of these six specimens). They concluded that some areas in South America exhibit elevatedTamanduamelanism frequency(from French Guiana to the Amazon basin and through to the eastern foothills of Ecuador and Peru), which could be related to speciation. However, our study clearly demonstrated that melanism is not a certainty implied in any speciation process as claimed by the previous authors.

Figure 6 Monmonier’s algorithm analysis to detect three most important geographical barriers for Tamandua sp. and Myrmecophaga tridactyla specimens following mitogenome sequencing Tamandua sp. (A) and Myrmecophaga tridactyla (B)
Gibb et al. (2016) also revealed very limited genetic differentiation between specimens ofT. mexicana(from Mexico) andT. tetradactyla(from French Guiana), with a genetic distance of only 2.8% for the mitogenome (2.1% at mtCOI). We found 4% divergence between MHI (mainly specimens with theT. tetradactylaphenotype) and MHII(mainly specimens with theT. mexicanaphenotype). These genetic distances typically represent internal population differentiation or incipient subspecies. Some morphological traits used to differentiateT. tetradactylaandT. mexicanainclude: (1) ear length (50-54 mm inT. tetradactylavs. 40-46 mm inT. mexicana), (2) pairs of orbital foramina (usually three inT. tetradactylavs. four inT. mexicana), (3) crescent completeness (incomplete on posterior border of infraorbital foramen inT. tetradactylavs. distinctly crescent shaped inT.mexicana; Wetzel, 1985), (4) number of caudal vertebrae(31-39 inT. tetradactylavs. 40-42 inT. mexicana, Wetzel 1975), and (5) body mass (3.4-8.0 kg inT. tetradactylavs.3.2-5.4 kg inT. mexicana,Wetzel, 1975). Therefore, species diagnoses inTamanduabased on differences in coat coloration, body size, skull characters, and number of caudal vertebrae can be quite variable within populations (Wetzel,1985), and thus have no systematic or phylogeographic value.
The karyotypes ofT. tetradactylaandT. mexicanaare also very similar. Both taxa have a diploid number (2n) of 54 chromosomes and a fundamental number (FN) of 104. The karyotype contains 16 metacentric and 10 submetacentric autosomal pairs. X is metacentric (submetacentric inT.mexicana), and Y is acrocentric (small submetacentric inT.mexicana) (Hsu, 1965; Hsu & Benirschke, 1969; Jorge et al.,1985; Jorge & Pereira, 2008; Pereira-Junior et al., 2004;Svartman et al., 2006). A previous study found a Brazilian specimen (which originated from the Atlantic rainforest in Sao Paulo State) with 56 chromosomes (Pereira-Junior et al.,2004), which was thus suggested as a potential new species.We did not sequence the mitogenome of any specimen from this region of Brazil and therefore cannot discard the existence of otherTamanduaspecies. Nevertheless, chromosomal polymorphism could occur within a species without the existence of different species (Ruiz-García et al., 2016).Therefore, our results are still consistent with the existence of a unique species within the genusTamandua, in which the three groups partially overlap.
The case ofM. tridactylais different. Traditionally, only one species has been considered as morphological differences have not been found in the different regions where this organism lives (Gaudin, 2018). Similarly, no karyotypic differences have been found among the different analyzed specimens ofM. tridactyla.Notably, Rossi et al. (2014)analyzed 26 Argentinian specimens of this species and obtained very similar results to those obtained by Hsu (1965)and Pereira-Junior et al. (2004) in different geographical areas. All specimens showed a chromosome number of 60(58+XX/XY; female/male) and an FN of 110. The only difference in the karyotypes of the 26 specimens was the morphology of chromosome pairs 2 and 3, which were heteromorphic in 65% of individuals. This polymorphism,involving the absence of small arms, is probably the result of pericentromeric inversion. Nonetheless, we detected very significant mitochondrial genetic differentiation between two groups of sympatricM. tridactyla, especially in northern South America (different areas of Peru and Amazon and Eastern Llanos of Colombia), although they were undifferentiated from a morphological and karyotypic point of view. This genetic differentiation was similar to or even greater than that found among the most divergent haplogroups ofTamandua,a prioriconsidered as different species. Thus, two cryptic subspecies or even semi-species may exist withinM. tridactyla. Recently,cryptic species have been identified for other Vermilingua taxa such asCyclopes(silky anteater). Miranda et al. (2018)recognized six new species of silky anteaters, instead of the single traditional species (C. didactylus), elevating three subspecies to species status, i.e.,Cyclopes dorsalis,Cyclopes catellus, andCyclopes ida, and describing three entirely new species, i.e.,Cyclopes xinguensis,Cyclopes thomasi, andCyclopes rufus. Thus, seven species are currently considered withinCyclopes. However, there is a fundamental difference in the systematics of possible cryptic species inCyclopesandMyrmecophaga. InCyclopes, these new molecularly detected species live in discrete and well-differentiated geographical areas, which has created systematically differentiated species.In contrast, theMyrmecophagagroups overlap in northern South America and are thus systematically challenging to differentiate in the field.
Where and when Tamandua and Myrmecophaga originated and possible subspecies
The original mitochondrial diversification ofTamanduaandMyrmecophaga, as well asCyclopes, seems to have occurred in different regions of South America. For the last genus,Coimbra et al. (2017) and Miranda et al. (2018) determined its origin in the southwestern Amazon (Rondonia and Inambari refuges in southern Peru and southwestern Brazilian Amazon). The northern Amazon, Atlantic Brazilian Forest, and Central American haplotypes originated later.
Herein, we showed that the oldest haplotypes inTamanduawere those from Uruguay. The haplotypes from Argentina,Paraguay, and Bolivia appeared later. The original mitochondrial diversification inTamanduaoccurred in southern South America, in one area outside the Amazon basin. Thus, the colonization ofTamanduawas from southern South America to northern South America and Central America.
In contrast,Myrmecophagaseems to have originated in the northwestern Amazon (current Peru and Colombia) and later colonized central and southern South America (in this case,we ignore the role of the Central AmericanMyrmecophaga).These results suggest that the processes of intra-generic diversification in the three extant Vermilingua genera were driven by different factors.
The fossil record for anteaters is highly incomplete due to their low diversity and difficult environmental conditions for fossilization. The earliest unquestioned anteater fossil can be dated to ~20 Mya in Patagonia (Carlini et al., 1992) during the Early Miocene (Colhuehuapian), although molecular estimates have dated the Cyclopedidae/Myrmecophagidae split to~38-46 Mya. Two other uncontroversial fossils are recognized, i.e.,Protamandua rothi(Ameghino, 1904) from the Early Miocene (Santacrucian), ~16 Mya (Delsuc et al.,2004), which has been considered a common ancestor to bothMyrmecophagaandTamandua(Hirschfeld, 1976), andPalaeomyrmidon incomptus(Rovereto, 1914) from the Late Miocene (Huayquerian) (Gaudin & Branham, 1998; McDonald et al., 2008), which has been considered a close relative of the silky anteater and of similar size, though it appears to have been entirely terrestrial. A third fossil genus,Neotamandua(Rovereto, 1914), has been recognized by some authors as strongly related to extantTamandua(Engelmann, 1985;Gaudin & Branham, 1998; Hirschfeld, 1976; McDonald et al.,2008), but other authors suggest it may be closely related(Patterson et al., 1992) or even congeneric toMyrmecophaga(McDonald et al., 2008). In fact, using morphological and molecular+morphological analyses, de melo Casali et al.(2020) consistently showed thatNeotamanduais closely related toMyrmecophaga. Additionally, based on clock analysis, de melo Casali et al. (2020) suggested thatNeotamanduais most likely part of an anagenetic line of descent leading toMyrmecophaga.
The times when these mitochondrial diversification processes began also differed for the three extant Vermilingua genera.Cyclopesapparently began diversification first.Coimbra et al. (2017) determined that this diversification began ~13.5 Mya (Middle Miocene), while Miranda et al.(2018) estimated that this process began ~10.3 Mya.Following the last authors, the split between the ancestor of the two original species ofCyclopesin the southwestern Amazon (C. rufusandC. thomasi) and the ancestor of the remainingCyclopesspecies (10 Mya) coincides with an increase in sedimentation rates in the Andean foreland basins that eventually overfilled, as well as a global sea level drop and climate cooling. Andean sediments reached the Atlantic coast through the Amazon drainage system, when the “Pebas”system changed into a fluvial “Acre” system (Hoorn et al.,2010).Myrmecophagawas the next genus to show mitochondrial diversification. Our estimates varied depending on the procedure used, i.e., ~6.2 Mya (Late Miocene,transition to Pliocene) with the BI procedure but 2.3 Mya(Pleistocene) with the MJN procedure. If we consider the first estimate valid, it coincides with the western Amazon wetlands~7-6 Mya. During this period, in the western Amazon,forested habitats and terrestrial conditions returned, which were correlated with an increase in plant diversity from 7-5 Mya. In addition, neotectonic activity (apparition of geological arches; Espurt et al., 2007, 2010; Patton et al.,2000; Patton & da Silva, 1998) and modification of fluvial systems were very important from ~7 to 3 Mya following the Paleogeography Hypothesis (Hoorn et al., 2010; Räsänen et al., 1995). In contrast, if the second estimate is accurate, this coincides with the final formation stage of the Andes,especially in the northern Andean area. At ~5 Mya, the northern Andes range was no more than 40% of its modernday elevation (Gregory-Wodzicki, 2000). Mountain uplift in this area between 5 and 2 Mya (Gregory-Wodzicki, 2000; Hoorn et al., 2010; Lundberg et al., 1998) may be a vicariant event responsible for the fragmentation and isolation of MHI, the firstMyrmecophagahaplogroup that originated ~3-2 Mya.
However, the paleontological data seem to favor the first estimate. An extinct anteater species from the Montehermosan South American Land Mammal Age (Late Miocene; 7-4 Mya),Nunezia caroloameghinoi(Kraglievich,1934), was later assigned toMyrmecophagaby McKenna &Bell (1997). The slightly olderNeotamandua magna(Ameghino, 1919) was transferred toNuneziaby Kraglievich(1934), and therefore should also be consideredMyrmecophaga. Thus, these two taxa represent the oldestMyrmecophagaand suggest that the genus extends back at least as far as the Huayquerian South American Land Mammal Age (Late Miocene, 9-7 Mya). This date is slightly younger but consistent with the molecular estimate of divergence betweenMyrmecophagaandTamandua,determined by Delsuc et al. (2004, 2012), Gibb et al. (2016),and de melo Casali et al. (2020) to have occurred ~10.1, 13.6,12.7, and 13.6 Mya, respectively. These dates are also consistent with the beginning of mitochondrial diversification of the current oldest haplotypes inMyrmecophagaat ~6 Mya.One hypothesis could relate the oldest haplotypes ofMyrmecophagafound in our study (MHI) with those present inM. caroloameghinoi(or a similar form; ~7-6 Mya), while the MHII haplotypes, which are more recent, could represent the actual form ofMyrmecophaga. This hypothesis agrees quite well with the results of de melo Casali et al. (2020), who demonstrated the existence of anagenesis in the branch ofNeotamanduaandMyrmecophaga. Indeed, the first known fossil undoubtedly belonging toM. tridactylawas from the Irvingtonian North American Land Mammal Age (Early Pleistocene, 1.9-1 Mya) in northern Mexico (Shaw &McDonald, 1987). Additional fossil material ofM. tridactylahas been collected from Late Pleistocene cave deposits in Brazil,and from Late Pleistocene sites in eastern Brazil and Uruguay(McDonald et al., 2008).
Tamanduaseems to be the most recent of the three extant Vermilingua genera, as also suggested by the fossil record.Fossils ofT. tetradactylawere formed during the Pleistocene in South America and during the Holocene in Central America(McDonald et al., 2008; McKenna & Bell 1997; Simpson,1945), although not specifically forT. mexicana(Siriano,1996). Our molecular estimates based on BI and MJN analyses were 1.8 Mya and 3.5-1.6 Mya, respectively. If the 3.5 Mya estimate is accurate, this agrees with the second estimate forMyrmecophaga. However, as its initial mitochondrial diversification was in southeastern South America, the last northern Andean uplift could not have been the cause of its diversification. When Gibb et al. (2016)applied the model of Condamine et al. (2013) to their data, the best temperature-dependent model showed that the speciation rate over the entire xenarthran time tree was negatively correlated with temperature. This pattern may be explained by several rapid speciation events in the xenarthran tree, especially within armadillos, which occurred in the last 15-5 million years during a period of intense cooling (Zachos et al., 2001), although these periods also occurred in the Pliocene to Pleistocene transition and within the Pleistocene.The continuous drop in temperature since the Middle and Late Miocene and in the Upper Pleistocene, followed by the current circum-Antarctic and last Andean uplift (Garzione et al., 2008),caused the aridification of South America and the formation of dry biomes such as the Chaco and Pampas in southern South America (Hoorn et al., 2010; Simon et al., 2009). Notably, the currentTamanduaoriginated in southern South America.
This higher temporal split could be related to the fact that we sampled moreTamanduaspecimens than previous studies estimating the split betweenT. tetradactylaandT.mexicana. We expect temporal splits to become older with denser taxon sampling (Gibb et al., 2016), which may have affected the coalescence among haplotypes found inTamandua. However, our estimates of 1.8-1.6 Mya(Pleistocene) were very similar to those obtained by Gibb et al. (2016) (1 Mya), Coimbra et al. (2017) (2.06 Mya), and de melo Casali et al. (2020) (1.6 Mya). The Gelasian period(2.5-1.8 Mya) was characterized by the last stages of a global cooling trend that led to the quaternary ice ages (International Commission on Stratigraphy, 2007). Both temperature (~4±2 °C lower) and precipitation (500-1 000 mm lower) were lower than present-day levels, with temperatures at 2 500 m a.s.l. up to 10 °C lower (Van der Hammen, 1992). Therefore,climatic change, more than geological changes, was the main cause ofTamanduadiversification. In fact,Tamanduadiversification seems to have occurred more rapidly than the colonization process inMyrmecophaga.
Our molecular results, with the first appearance of the ancestor ofCyclopes, followed by the ancestor ofMyrmecophaga, and later by the ancestor ofTamandua,agree well with the karyotype evolution of Vermilingua.Cyclopeshas a karyotype of 2n=64, whereasMyrmecophagaandTamanduahave karyotypes of 2n=60 and 2n=54,respectively. A mechanism of reversible fusion/fission and reciprocal translocation was proposed by Jorge et al. (1985) to explain the reduction in chromosome number from 64 to 54 in Vermilingua. This correlated well withCyclopes, the oldest,andTamandua, the youngest.
On the other hand, withinTamandua, three significant groups were detected, as mentioned above. Internal diversification within these three haplogroups was relatively similar (THI: 0.9 Mya (MJN)-1.2 Mya (BI); THII: 0.6 Mya(MJN)-0.7 Mya (BI); THIII: 0.7 Mya (MJN)-1.3 Mya (BI)) and all diversified during the Pleistocene. Indeed, the diversification haplotype times (1.3-0.8 Mya) corresponded to the Pre-Pastonian glacial period, which was the highest glacial peak of the first Quaternary glacial period (Günz). This glacial period was extremely dry, and there was a high degree of forest fragmentation. It was a time for haplotype diversification in many mammals, e.g., Pampas cat,Leopardus colocolo(Cossíos et al., 2009), Andean fox,Lycalopex culpaeus(Ruiz-García et al., 2013), jaguarundi,Puma yagouroundi(Ruiz-García et al., 2018b, 2018c), and woolly monkey from the Peruvian and Bolivian Andes,Lagothrix lagothricha tschudii(Ruiz-García et al., 2019). Our analyses clearly showed that THI (from southern-eastern South America) gave way to THII in northern South America (basically current Colombia and Ecuador). This occurred ~1.1 Mya (MJN) or ~1.6 Mya (BI).Therefore, our results support two possible hypotheses: (1) “in situ” in northern South America, THII gave way to THIII (from MJN ~0.8-0.7 Mya), which was initially an isolated and possibly very fragmented population. In this case, their genetic distances were highest (10%-11%) in reference to the other two haplogroups due to founder effects. Additionally, THIII had the greatest nucleotide diversity. Later, THII gradually colonized Central America (absence of clear population expansions in our results for this group). In parallel, or prior to the entrance of THII in Central America, THIII strongly expanded and colonized a large proportion of what would eventually be Colombia and a small fraction of Ecuador and colonized Central America (we detected significant population expansions for this haplogroup, sometimes older than those for THII). If this hypothesis occurred, then Central America was independently colonized twice (THII and THIII) byTamandua. (2) Once THII appeared in northern South America, it gradually colonized Central America, and “in situ”in Central America, THII gave rise to THIII, which, in turn,explosively colonized northern North America again (current Colombia and a small area of Ecuador). The haplotypes of THIII geographically overlapped the haplotypes of THI and THII previously present in northern South America. Clearly,THIII (with bothTamanduaphenotypes at near 50%) is not a hybrid group of THI (mainlyT. tetradactylaphenotypes) and THII (mainlyT. mexicanaphenotypes); if it were, we would expect an intermediate genetic distance of ~2% from both THI and THII, which was not the case (10%-11%).
These new results dramatically change our view of the colonization of Central America byTamandua. For instance,Moraes-Barros & Arteaga (2015), Gibb et al. (2016), and Coimbra et al. (2017) considered that the previous estimate of 2 Mya betweenT. tetradactylaandT. mexicanawell matched the ending of the final uplift of the northern Andes (5-2 Mya;Hoorn et al., 2010; Lundberg et al., 1998), and that this geological event may explain the vicariance detected between the twoTamanduaspecies. We now know that the northern Andes probably caused the apparition of THII and THIII (and certain fragmentation within THIII) but was not a barrier forTamanduabecause at least two colonization events occurred in Central America (or one colonization event from northern South America to Central America and another colonization event from Central America to northern South America). If this vicariance event could be invalidated, this would strengthen the existence of a single species ofTamandua. Additionally,some authors (Abba & Superina, 2010; Moraes-Barros &Arteaga, 2015; Superina et al., 2010; Wetzel, 1975, 1985)consider that the traditional model ofTamandua, i.e.,speciation after isolation on both sides (west and east) of the Andes, is comparable to the cases ofCabassous centralisandCabassous unicinctus. Our results indicate that the case ofTamanduais not necessarily comparable with the situation found in other species. For instance, Ruiz-García et al.(2018a, 2020) estimated a strong temporal divergence between the trans- and cis-AndeanBradypus variegatuspopulations at ~13.2-9.7 Mya, i.e., during the Miocene, which was not the case forTamandua. Ruiz-García et al. (2018b)also suggested that each species exhibited a different capacity for colonization. Furthermore, Gibb et al. (2016)showed a fair degree of branch length heterogeneity among groups in their xenarthran mitogenomic tree, with fast evolving clades, including anteaters and Dasypodinae, and slow evolving clades, such as sloths and hairy armadillos. One possible similarity may exist betweenTamanduaand xenarthranD. novemcinctus. During expansion of its range,the nine-banded armadillo was part of at least two dispersal waves from South America to North America (Arteaga et al.,2012). This was reflected in the presence of two divergent lineages that arose in Central America and Mexico and moved into Colombia.
The utility of geographical subspecies—in the sense of Mayr(1942, 1963)—is limited for the uniqueTamanduaspecies detected in this study as the three haplogroups overlapped,e.g., in Colombia and Ecuador. However, there should be partial correspondence among certain subspecies previously described for bothT. tetradactylaandT. mexicanaand the“new”T. tetradactyla. Here, two previously nominated subspecies,T. t. nigra(Guyana) andT. t. tetradactyla(Pernanbuco, Brazil), were not sampled and thus we cannot infer anything about them. In addition,T. t. quichua(San Martín, Peru) was not differentiated from other taxa and therefore we negate its validity. However, there may be unique correspondence betweenT. t. straminea(southern South America) and THI (also from southern and central South America). RegardingT. mexicana, the geographical area ofT.m. mexicana(Tabasco, Mexico) was not sampled and we cannot infer anything about its systematics. In addition,T. m.punensis(Guayas, Ecuador) was intermixed in different haplogroups and we negate its validity. However,T. m.opistholeuca(Colombia and Central America) andT. m.instabilis(northern Colombia and Venezuela) could be related to THII and THIII, respectively. Hence, we propose one unique species,T. tetradactyla, with three possible subspecies (at least in the geographical area studied): i.e.,T. tetradactyla tetradactyla(THI),T. tetradactyla opistholeuca(THII), andT.tetradactyla instabilis(THIII).
Traditionally, three subspecies ofM. tridactylahave been considered. As commented forTamandua, the utility of subspecies inMyrmecophagais limited due to the geographical overlapping of the different haplogroups. AsM. t.centraliswas not sampled, we cannot provide any systematic inference. However, the most northern group (MHI) could be related toM. t. artata, and the most expanded South American group (MHII) could be related toM. t. tridactyla.
Genetic diversity, population changes, and spatial genetic structure within Tamandua and Myrmecophaga
BothTamanduaandMyrmecophagashowed elevated levels of genetic diversity, indicating the long existence of these taxa in the Neotropics. Thus, at a macro-geographical level, both anteater species seem to have historically large population sizes.
Both taxa also showed evidence of female population expansions, although they were stronger inTamanduathan inMyrmecophaga. For the first genus, both mismatch distribution and BSP showed population increase within the range of~0.5-0.17 Mya. The second genus only showed evidence of population increase for BSP ~0.25 Mya. This period coincides with the last phase of the Bonaerense Stage (Ameghino,1889) from 0.5 to 0.13 Mya and is characterized by a large increase in mammals with Holarctic origin in South America.This was a time of stable climate, with a warm and humid environment, which could favor population expansions,especially ofTamandua. However, for both anteater genera, a strong female population decrease was detected around 10 000-20 000 years ago. This agrees well with a major extinction period, which seems to have preferentially affected the largest terrestrial forms, such as giant ground sloths, glyptodonts, and saber toothedSmilodon(Lessa et al., 1997; Lyons et al.,2004). Indeed, this massive extinction was caused by an extremely cold period (11 000-10 000 years ago). This period(named “El Abra” in the northern Andes, Younger Dryas or Dryas III in Scandinavia and northern Europe, 11 800-9 600 years ago (Clapperton, 1993), or central European Tardiglacial (Dollfus, 1964)) was crucial in the extinction of mammals. Indeed, 80% of large mammal species (around 40 species) were eliminated from North America (Martin, 1989;Pielou, 1991).
The spatial genetic structures of both anteater genera were not strong because the different haplogroups geographically overlapped across a broad area. However, in some analyses(e.g., AIDA),Tamanduashowed an isolation-by-distance pattern. For BrazilianTamandua, Clozato et al. (2015)detected some positive and significant correlations between genetic and geographic distances forHLA-DRBand microsatellite markers, although the degree of correlation was not very high (DRB: r=0.081,P=0.002; microsatellites:r=0.164,P=0.001). With microsatellites, Clozato (2014)claimed that the Brazilian Amazon had the greatest genetic differentiation and highest genetic diversity for all Brazilian populations ofT. tetradactylastudied and concluded that this was the center of diversification for the species. Nevertheless,our results clearly showed that the Amazon was not the origin center ofTamandua. Clozato (2014) did not reach this conclusion because they did not consider two aspects. First,they only sampledTamanduasin Brazil and this species has a considerably broader geographical distribution from Mexico to northern Argentina and Uruguay, and second although the three haplogroups ofTamanduaconverged in the northern Amazon, they did not originate there.
ForMyrmecophaga, Clozato et al. (2017) did not detect significant spatial genetic structure in Brazil, as we did in South America. One biological condition that could help to create inexistent spatial genetic structure in the giant anteater is the relatively high dispersion capacity, at least, of males.Males with adjacent home ranges show low levels of relatedness (Prodöhl et al., 2008). Clozato et al. (2017)detected the Amazon population as the most differentiated one, although it was not differentiated from theMyrmecophagapopulation from the Atlantic Forest. We did not sampleMyrmecophagain the Brazilian Atlantic Forest.The Amazon and Atlantic Forest biomes are separated by a large distance, with intermediate dry territories (Caatinga,Cerrado, Pantanal, and Chaco). However, the Cerrado has several fragments of deciduous and semi-deciduous forests,as well as gallery forests that create a bridge between the Amazon and Atlantic forests (Costa, 2003; Vivo, 1997).Moreover, both rainforests were possibly continuous in the past, and this old connection may have encouraged interchange of specimens of different taxa between the two biomes (Bigarella et al., 1975; Costa, 2003; Martins et al.,2007). Thus, the Brazilian Atlantic Forest probably does not contain aMyrmecophagahaplogroup different from the two detected in the current study.
Here, we present the first mitogenome study at a continental scale forTamanduaandMyrmecophaga, showing three main mitogenome groups inTamanduaand two inMyrmecophaga.However, additional nuclear genomes should be analyzed to complement the current mitogenome analysis. Furthermore,several other geographical areas should be studied with dense sampling, including the Guianas for both taxa, many areas of Brazil forTamandua(with mitogenomes), and Central America forMyrmecophaga. The study of this last taxon is urgent as it is a vulnerable species, which has become extinct in many geographical areas.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
T he authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
M.R.G. designed and obtained a large proportion of the samples for the study. O.M.G., C.M.P., and J.B. obtained some samples. M.R.G. and J.M.S. supervised the molecular analyses. D.P.B. performed the laboratory procedures. M.R.G.performed the statistical analyses and wrote the manuscript with input from the other authors. M.R.G. and D.P.B.submitted the sequences to GenBank. O.M.G., C.M.P., J.B.,and J.M.S. revised the manuscript. All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Diana Alvarez, Pablo Escobar-Armel, Nicolás Lichilín, and Luisa Fernanda Castellanos-Mora for their respective help in obtainingTamanduaandMyrmecophagasamples over the last 20 years. We thank Dr. Enrique Richard and Dr. Juan Pablo Juliá for providing samples ofMyrmecophagafrom Argentina. We also thank the Ministerio del Ambiente Ecuatoriano (MAE) in Santo Domingo de Tsáchilas and Coca, Instituto von Humboldt (Colombia),Peruvian Ministry of Environment, PRODUCE (Dirección Nacional de Extracción y Procesamiento Pesquero), Consejo Nacional del Ambiente and the Instituto Nacional de Recursos Naturales (INRENA) from Peru, Colección Boliviana de Fauna(Dr. Julieta Vargas), and CITES Bolivia for their roles in facilitating the collection permits in Ecuador, Colombia, Peru,and Bolivia. The first author also thanks the many people of the diverse Indian tribes in Ecuador (Kichwa, Huaorani, Shuar,and Achuar), Colombia (Jaguas, Ticunas, Huitoto, Cocama,Tucano, Nonuya, Yuri, and Yucuna), Peru (Bora, Ocaina,Shipigo-Comibo, Capanahua, Angoteros, Orejón, Cocama,Kishuarana, and Alamas), and Bolivia (Sirionó, Canichana,Cayubaba, and Chacobo) for their assistance in obtainingTamanduaandMyrmecophagasamples, as well as the diverse Mayan communities and people from Guatemala,Honduras, and Panama for their assistance in obtainingTamanduasamples.

- Zoological Research的其它文章
- Chromosome-scale genome assembly of brownspotted flathead Platycephalus sp.1 provides insights into demersal adaptation in flathead fish
- Captopril alleviates lung inflammation in SARS-CoV-2-infected hypertensive mice
- Role of juvenile hormone receptor Methoprene-tolerant 1 in silkworm larval brain development and domestication
- Deletion of phosphatidylserine flippase β-subunit Tmem30a in satellite cells leads to delayed skeletal muscle regeneration
- Global view on virus infection in non-human primates and implications for public health and wildlifeconservation
- Prosecution records reveal pangolin trading networks in China, 2014-2019